UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
|
☒ |
Annual Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the fiscal year ended January 2, 2021
or
|
☐ |
Transition Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
Commission File No. 000-19621
JANONE INC.
(Exact name of registrant as specified in its charter)
|
Nevada |
|
41-1454591 |
|
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
|
|
|
|
|
325 E. Warm Springs Road, Las Vegas, Nevada |
|
89119 |
|
(Address of principal executive offices) |
|
(Zip Code) |
Registrant’s telephone number, including area code: 702-997-5968
Securities registered pursuant to Section 12(b) of the Act:
|
Common Stock, $0.001 par value Title of each class |
JAN Trading Symbol(s) |
NASDAQ Capital Market Name of each exchange on which registered |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☐ Yes ☒ No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. ☐ Yes ☒ No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. ☒ Yes ☐ No
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ☒ Yes ☐ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer |
☐ |
|
Accelerated filer |
☐ |
Non-accelerated filer |
☒ |
|
Smaller reporting company |
☒ |
|
Emerging growth company |
☐ |
|
|
If any emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal controls over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). ☐ Yes ☒ No
The aggregate market value of the registrant’s common stock held by non-affiliates, based on the closing sales price of such stock on June 27, 2020 was $7,153,000.
The number of shares outstanding of the registrant’s common stock as of March 25, 2021 was 2,403,410.
TABLE OF CONTENTS
|
|
|
Page |
||
|
|
|
|
||
|
|
|
|
|
|
|
Item 1. |
|
|
1 |
|
|
Item 1A. |
|
|
38 |
|
|
Item 2. |
|
|
53 |
|
|
Item 3. |
|
|
53 |
|
|
Item 4. |
|
|
53 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 5. |
|
Market for Our Common Equity, Related Shareholder Matters and Issuer Purchases of Equity Securities |
|
54 |
|
Item 6. |
|
|
54 |
|
|
Item 7. |
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
|
55 |
|
Item 7A. |
|
|
62 |
|
|
Item 8. |
|
|
63 |
|
|
Item 9. |
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
|
64 |
|
Item 9A. |
|
|
64 |
|
|
Item 9B. |
|
|
65 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 10. |
|
|
66 |
|
|
Item 11. |
|
|
69 |
|
|
Item 12. |
|
Security Ownership of Certain Beneficial Owners and Management and Related Shareholder Matters |
|
72 |
|
Item 13. |
|
Certain Relationships and Related Transactions, and Director Independence |
|
74 |
|
Item 14. |
|
|
75 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Item 15. |
|
|
76 |
|
|
Item 16. |
|
|
76 |
|
|
|
|
|
|
|
|
|
77 |
|||
|
|
80 |
|||
i
General
As of September 10, 2019, JanOne Inc. (formerly known as Appliance Recycling Centers of America, Inc.) and subsidiaries (collectively, “we,” the “Company,” or “JanOne”) changed its name and broadened its business perspectives to being a pharmaceutical company focused on finding treatments for conditions that cause severe pain and bringing to market drugs with non-addictive pain-relieving properties. The Company aims to reduce prescriptions for dangerous opioid drugs by treating underlying diseases that cause severe pain. Our first drug candidate is a treatment for Peripheral Arterial Disease (“PAD”), a condition that can cause severe pain and affects over 8.5 million people in the U.S. alone. From digital technologies to educational advocacy to revolutionary painkilling drugs that address what we believe is a multibillion dollar a year market, the Company intends to champion new initiatives to help combat the opioid crisis, which claims tens of thousands of lives each year. The new name, JanOne, was strategically chosen to express the start of a “new day” in the fight against the opioid epidemic. January First is the first day of a New Year—a day of optimism, resolution, and hope. JanOne affirms the Company’s new strategic commitment to fresh thinking and innovative means to assist in ending the worst drug crisis in our nation’s history. The Company also adopted a new Nasdaq ticker symbol, NASDAQ: JAN, a new CUSIP number, 03814F403, and a new website address – www.janone.com.
We continue to operate our legacy businesses, ARCA Recycling, Inc. (“ARCA Recycling”) and Customer Connexx, LLC (“Connexx”), a company that provides call center services for recycling business, in our Recycling segment, and GeoTraq Inc. (“GeoTraq”), in our Technology segment. ARCA Recycling recycles major household appliances in North America by providing turnkey appliance recycling and replacement services for utilities and other sponsors of energy efficiency programs. GeoTraq is engaged in the development, design, and, ultimately, we expect, the sale of cellular transceiver modules and associated wireless services.
The information contained in or accessible from our website is not incorporated into this Annual Report on Form 10-K (the “Form 10-K”), and you should not consider it part of this Form 10-K. We have included our website address in this Form 10-K solely as an inactive textual reference.
We were incorporated in Minnesota in 1983, although, through our predecessors, we began operating our legacy recycling business in 1976. On March 12, 2018, we reincorporated in the State of Nevada. Our principal office is located at 325 E. Warm Springs Road, Suite 102, Las Vegas, Nevada 89119.
Recent Developments
On February 19, 2021, we, together with our subsidiaries (a) ARCA Recycling and (b) Connexx, entered into an Asset Purchase Agreement (the “Purchase Agreement”) with (i) ARCA Affiliated Holdings Corporation, a Delaware corporation, (ii) ARCA Services Inc., a Delaware corporation, and (iii) Connexx Services Inc, a Delaware corporation (collectively, the “Buyers”), pursuant to which the Buyers agreed to acquire substantially all of the assets, and assume certain liabilities, of ARCA and Connexx (the “Disposition Transaction”). The principal of the Buyers is Virland A. Johnson, our Chief Financial Officer. The Disposition Transaction is expected to be consummated on or before August 18, 2021. In the event the Disposition Transaction is not closed by such date, the Purchase Agreement may be terminated and, in accordance with its terms, the Buyers may be required to pay to us a “break fee” of $250,000. The Purchase Agreement and the Disposition Transaction were unanimously approved by our Board of Directors at a meeting during the portion of which the Purchase Agreement and Disposition Transaction were considered and voted on while Mr. Johnson was not present. The purchase price that the Buyers have agreed to pay to us in the Disposition Transaction is $25.0 million, subject to certain adjustments, including a potential increase in the purchase price due to an earnout, the assumption of certain debt of ARCA, Connexx, or us, and potential indemnification claims (collectively, the “Initial Aggregate Consideration”). At closing, $7.5 million of the Aggregate Consideration will be paid in immediately available funds, and $17.5 million of the Initial Aggregate Consideration will be paid pursuant to the terms of the Buyers’ promissory note in our favor (the “Note”), which Note will bear interest at the rate of 6% per annum on the unpaid balance thereof. The Buyers’ payment obligations under the Note will be subordinated to the Buyers’ obligations to their Disposition Transaction lender(s),
1
with the terms of such subordination to be determined upon Buyers’ identification of their lender(s). The parties have made customary representations, warranties, covenants, and indemnities in connection with the Disposition Transaction. Commencing on February 19, 2021, (i) the Buyers will seek financing for the balance of the Initial Aggregate Consideration and (ii) the parties will prepare and negotiate the terms and conditions of certain ancillary documentation, including, without limitation, disclosure schedules, bills of sale, assignment and assumption agreements, the Note, and any related subordination documentation with Buyers’ Disposition Transaction lender(s).
The Purchase Agreement contains certain representations and warranties that the parties made to each other as of the date of the Purchase Agreement or such other date as specifically referenced therein. The representations and warranties were made solely for purposes of the Purchase Agreement and (i) are subject to limitations agreed by the parties in negotiating the terms and conditions thereof, (ii) may not be accurate or complete as of any specified date, (iii) will be qualified by the underlying disclosure schedules, (iv) may be subject to a contractual standard of materiality different from those generally applicable to investors, and (v) may have been used for the purpose of allocating risk among the parties thereto, rather than for establishing any matters as facts. Information concerning the subject matter of the representations and warranties may change after February 19, 2021, which subsequent information may or may not be fully reflected in JanOne’s public disclosures. For the foregoing reasons, the representations and warranties contained in the Purchase Agreement should not be relied upon as statements of factual information.
The foregoing descriptions of the Purchase Agreement and the Disposition Transaction do not purport to be complete and are qualified in their entirety by reference to the Purchase Agreement, a copy of which is filed as Exhibit 2.3 to this Form 10-K and is incorporated herein by reference.
Biotechnology
Overview
We are a clinical-stage biopharmaceutical company focused on becoming the leader in identifying, acquiring, licensing, developing, partnering, and commercializing novel, non-opioid and non-addictive therapies to address the large unmet medical need for the treatment of pain. Our initial product candidate, JAN101 (formerly known as TV1001SR), is a potential treatment for PAD, a vascular disease that affects more than 8.5 million people in the U.S. and more than 60 million people worldwide. We expect to commence Phase 2b clinical trials for the treatment of PAD in 2021. We are also researching the potential impact our compound JAN101 could have in patients with COVID-19 as many doctors around the world and our company believes COVID-19 is a respiratory disease that directly affects the vascular system. In March 2021, we determined to defer our current, on-going pursuit of JAN101 as a potential treatment for COVID-19 vascular complications. In connection with that determination, we will not currently file a utility patent nor submit an investigational new drug application (“NDA”) to the FDA. This determination was based on several factors, including the current, positive effect that three vaccines are having on the COVID-19 pandemic in the United States. Should the vaccines ultimately prove less effective than currently expected or should new COVID-19 variants result in another pandemic, we may then determine to end our deferral of pursuing JAN101 as a treatment of COVID-19 vascular complications.
2
JAN101
Generally
JAN101, formerly known as TV1001SR, our advanced product candidate, is a patented oral, sustained release pharmaceutical composition of sodium nitrite and targets poor blood flow to the extremities, such as those with vascular complications of diabetes or PAD and treats pain. A conclusion from a round of human studies found JAN101 prevents the prevalent reports of headaches by patients treated with an immediate release formulation of sodium nitrite. In a previous study of patients with PAD, 40 mg BID treatment with immediate release sodium nitrite led to a statistically significant reduction in reported pain while a 80 mg BID treatment had the more pronounced effect on bioactivity and Flow Mediated Dilation, a measure of vascular function. However, a number of subjects on both treatment groups reported headaches and dizziness following treatment. Although this did not result in subjects discontinuing treatment, JAN101 was developed to overcome this side effect. JAN101 was tested in a bridging study of diabetic neuropathy subjects and during that bridging study, the subjects did not report headaches or dizziness. Subjects in this bridging study also reported less pain following treatment and improvements in bioactivity (quantitative sensory testing, a measure of nerve function) were similar to the PAD study, where the 80 mg dose group had the greatest improvement in Flow Mediated Dilation. The ability to alleviate pain with BID treatment of JAN101 offers promise for a new non-addictive, non-sedating treatment of chronic pain.
Clinical Studies in Humans JAN101 Attributes
|
|
• |
Well established safety profile |
|
|
• |
Excellent bioavailability |
|
|
• |
Lack of induced tolerance |
|
|
• |
Non-narcotic |
JAN1010 does not mask pain, but instead treats the cause of pain by improving tissue and vascular function.
Benefits of Sodium Nitrite on Vascular Health
In initial research studies, sodium nitrite effectively restored ischemic tissue blood flow and was effective in a wide range of pathologies involving alterations of angiogenesis - development of new blood vessels - including diabetes, wound healing and tissue necrosis. Beneficial effects included enhancing angiogenesis, endothelial cell proliferation, and arteriogenesis. There is also a strong association between reduced circulating nitrite levels and cardiovascular diseases in humans. We describe some of the associations and beneficial effects of sodium nitrite/nitrite below.
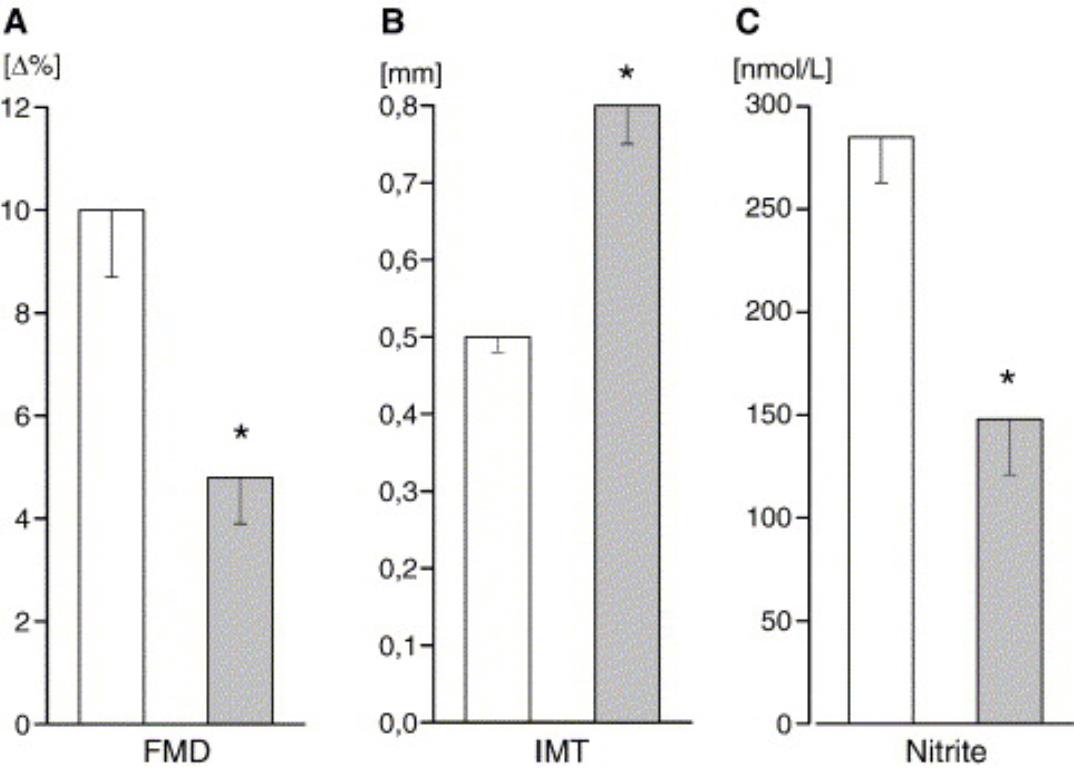
Plasma nitrite levels are negatively correlated to cardiovascular disease
3
Plasma nitrite levels were inversely related to number of cardiovascular risk factors a subject had and decreased plasma nitrite was associated with decreased flow mediated vasodilation (FMD) and increased intimal medial thickness (IMT) (both indicators of vascular pathology). -Kleinbongard, et al. (2006) Free Radic Biol and Medicine 40:295-302.
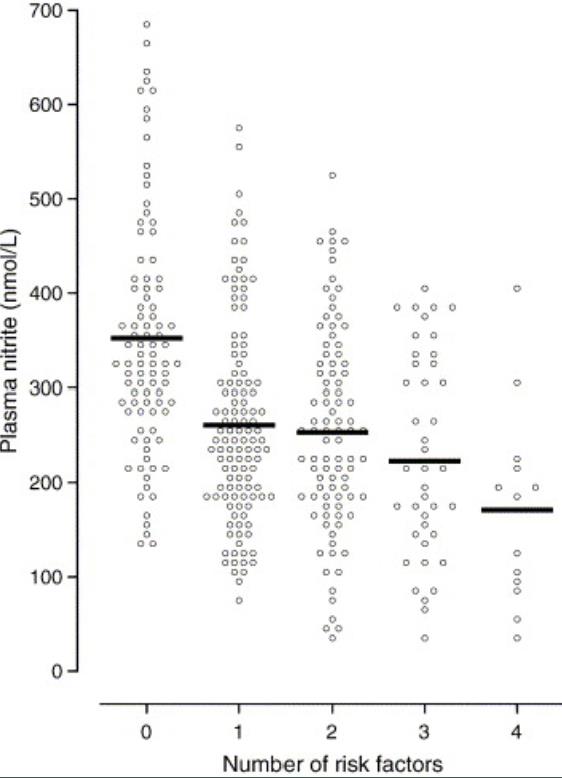
Plasma nitrite levels are reduced in diabetic and PAD patients
Exercise is a well-known stimulator of endothelial nitric oxide synthase activity, NO production that leads to increased plasma nitrite. In the study by Allen et al, these authors revealed that baseline plasma levels of nitrite were less in patients with diabetes mellitus (DM) or DM + PAD. Importantly, increases in plasma nitrite levels were not observed in either DM, PAD or DM + PAD patients after supervised exercise. These data reveal that baseline nitrite availability is compromised in DM patients and that supervised exercise is unable to increase plasma nitrite levels but actually results in a decrease in nitrite highlighting a physiological efficiency of this molecule. -Allen et al Nitric Oxide 2009 20:231-237.
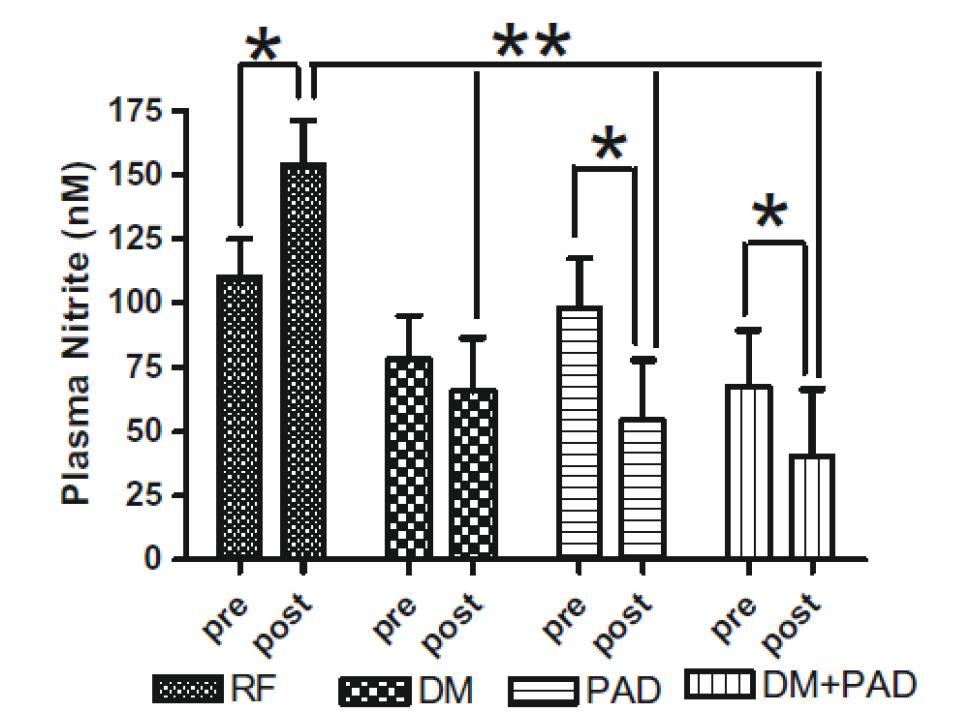
Skeletal Muscle Nitrite and Metabolite Levels are Reduced in Critical Limb Ischemia Patients
4
Skeletal muscle nitrite, nitrosothiol, nitric oxide-heme and cGMP are all significantly reduced in CLI patients. Diabetic patients with CLI show even further nitrite reductions.
In summary, nitrite levels in various cardiovascular and vascular diseases appear to be inversely related to the severity of the disease in humans:
|
|
• |
Lower nitrite levels are associated with higher level of heart failure; |
|
|
• |
Lower nitrite levels are observed in diabetic patients with PAD and are not compensated by exercise; and |
|
|
• |
Nitrite levels are lower in the muscles of patients with critical limb ischemia and are further reduced in diabetic subjects with critical limb ischemia. |
Given the association between low levels of circulating nitrite and human diseases, supplementation with sodium nitrite has been studied preclinically in animals. Below are summaries of some of the more important findings:
|
|
• |
Promotes angiogenesis |
|
|
• |
Stimulates wound healing |
|
|
• |
Prevents tissue necrosis |
5
From Arya et al.
Nitrite Therapy Selectively Increases Ischemic Tissue Vascular Density in a NO-dependent Manner
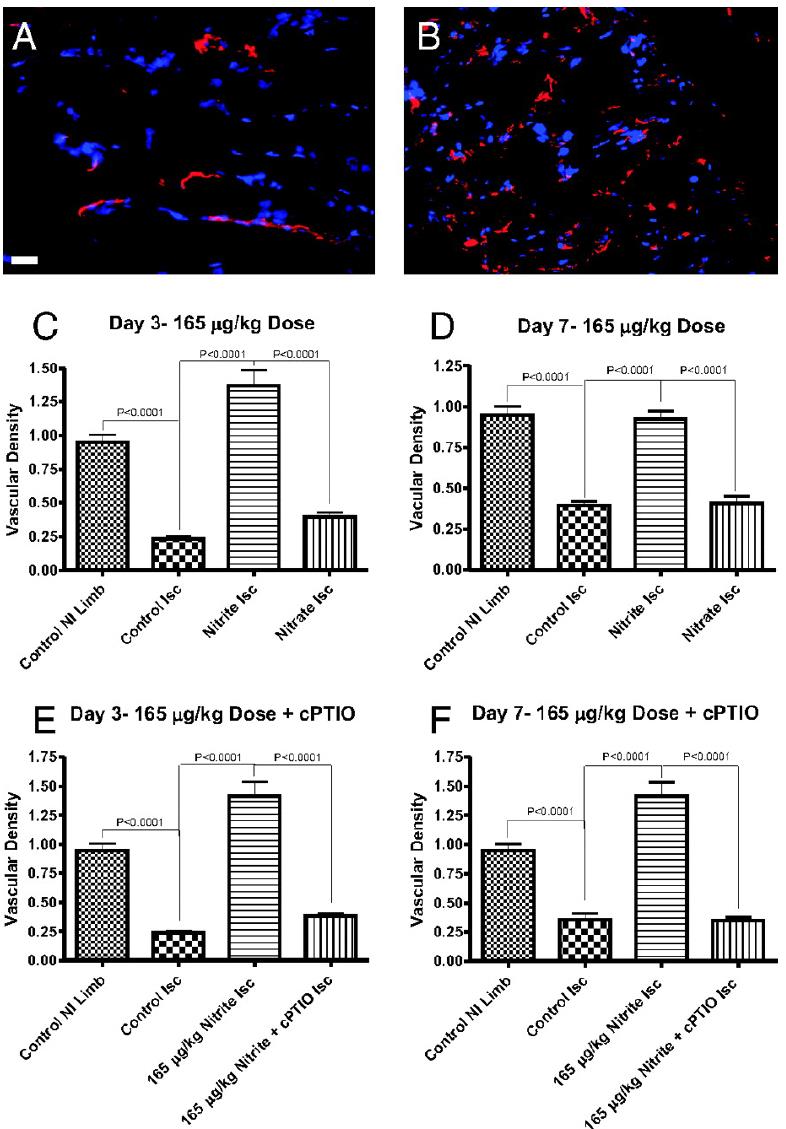
Chronic sodium nitrite therapy increases ischemic tissue vascular density in a NO-dependent manner. A and B show representative images of CD31 (red) and DAPI nuclear (blue) staining from sodium nitrite and sodium nitrate ischemic gastrocnemius muscle tissue at day 7. C and D report the vascular density of ischemic gastrocnemius muscle tissue at days 3 and 7 for 165 μg/kg sodium nitrite and nitrate treatments, respectively. E and F demonstrate the vascular density of ischemic gastrocnemius muscle tissue at days 3 and 7 from 165 μg/kg sodium nitrite plus carboxy PTIO. (Scale bar, 150 μm.) n = 10 mice per treatment group. Kumar D. et al. PNAS; 2008; 105:7540-7545.
6
Nitrite Therapy Augments Arterial Perfusion of Ischemic Tissue
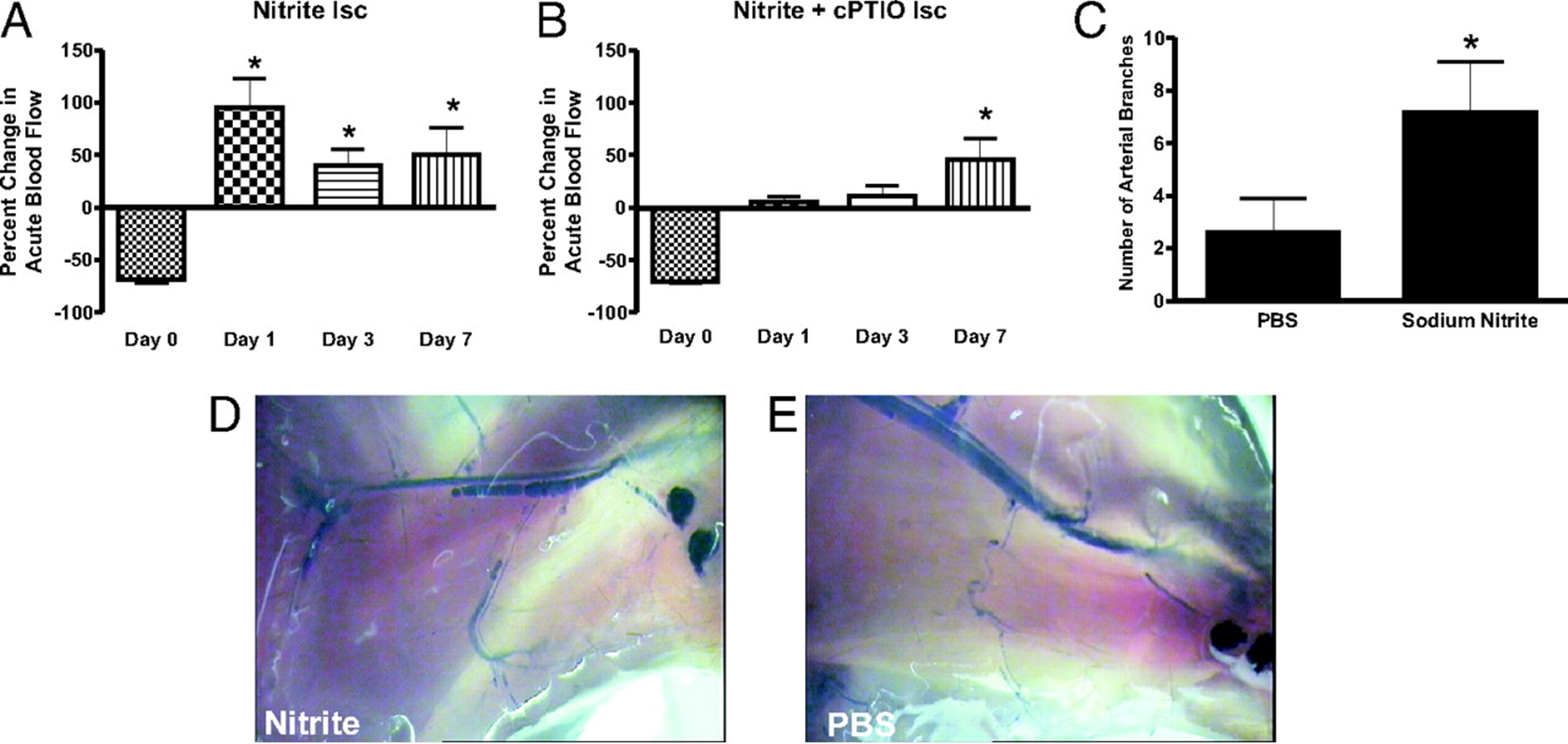
Chronic sodium nitrite therapy acutely increases ischemic tissue blood flow and stimulates arteriogenesis. A and B report 165 μg/kg sodium nitrite-induced acute changes in blood flow of chronically ischemic tissues at various time points with or without cPTIO, respectively. C reports the number of arterial branches between PBS and nitrite therapies. D and E illustrate vascular casting of the arterial vasculature in ischemic hind limbs of day 7 nitrite or PBS-treated mice, respectively. *, P < 0.01 vs. sodium nitrate. n = 10 mice per treatment group. Kumar D. et.al. PNAS;2008; 105:7540-7545.
7
Nitrite Therapy Restores Diabetic Ischemic Hind-Limb Blood Flow and Promotes Wound Heal
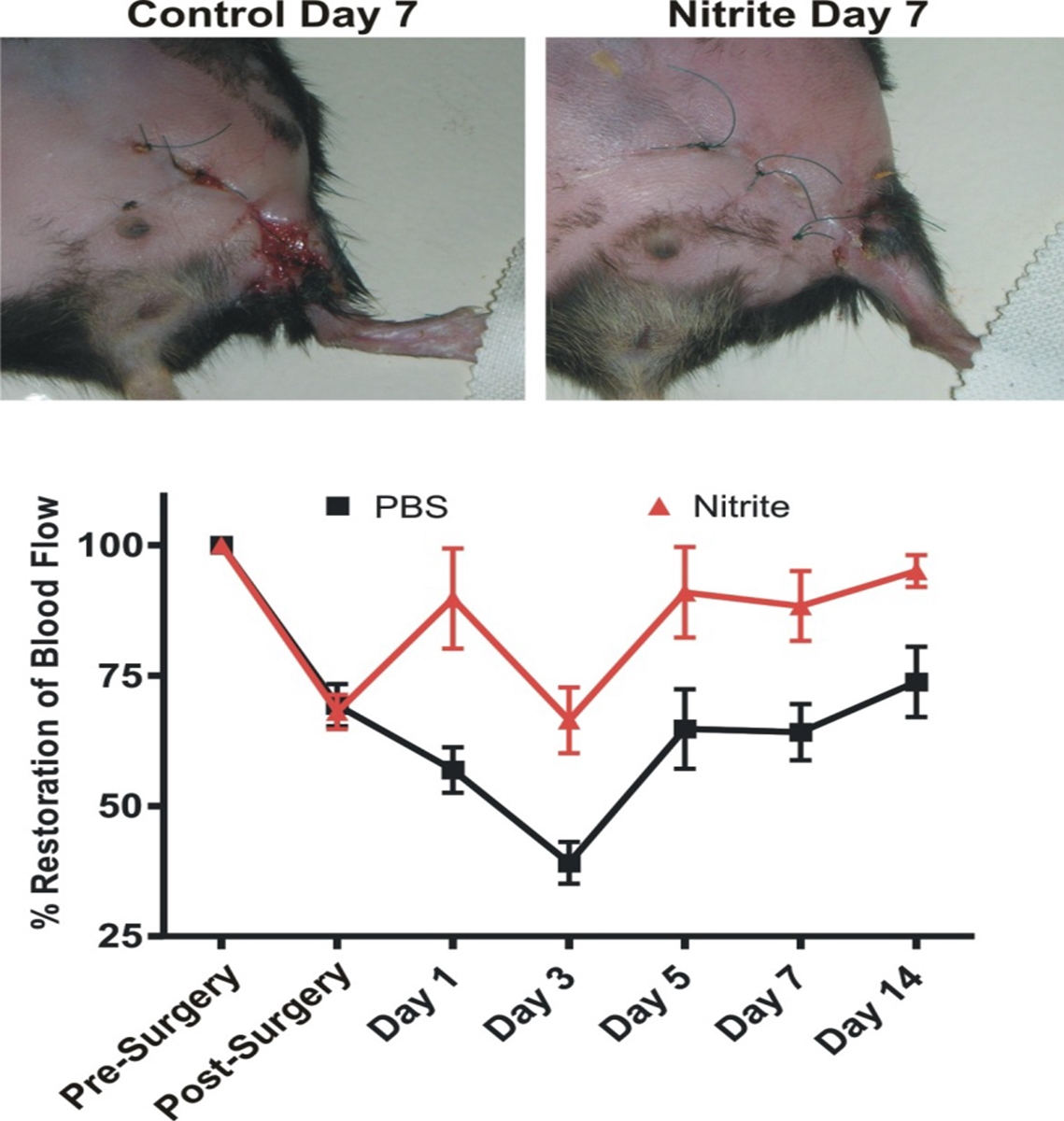
Unilateral femoral artery ligation was performed on 18-20 week old male Db/Db mice. Mice were randomized to PBS or sodium nitrite (165 μg/kg) therapy twice daily via I.P. injection. Laser doppler flowmetry was performed at the indicated time points. Increased wound dehiscence was noted in the PBS treated animals at day 7 but not in nitrite treated animals. (Bir et al Diabetes 2014, 63(1):270-81).
Nitrite Therapy Increases Diabetic Ischemia Induced Angiogenesis
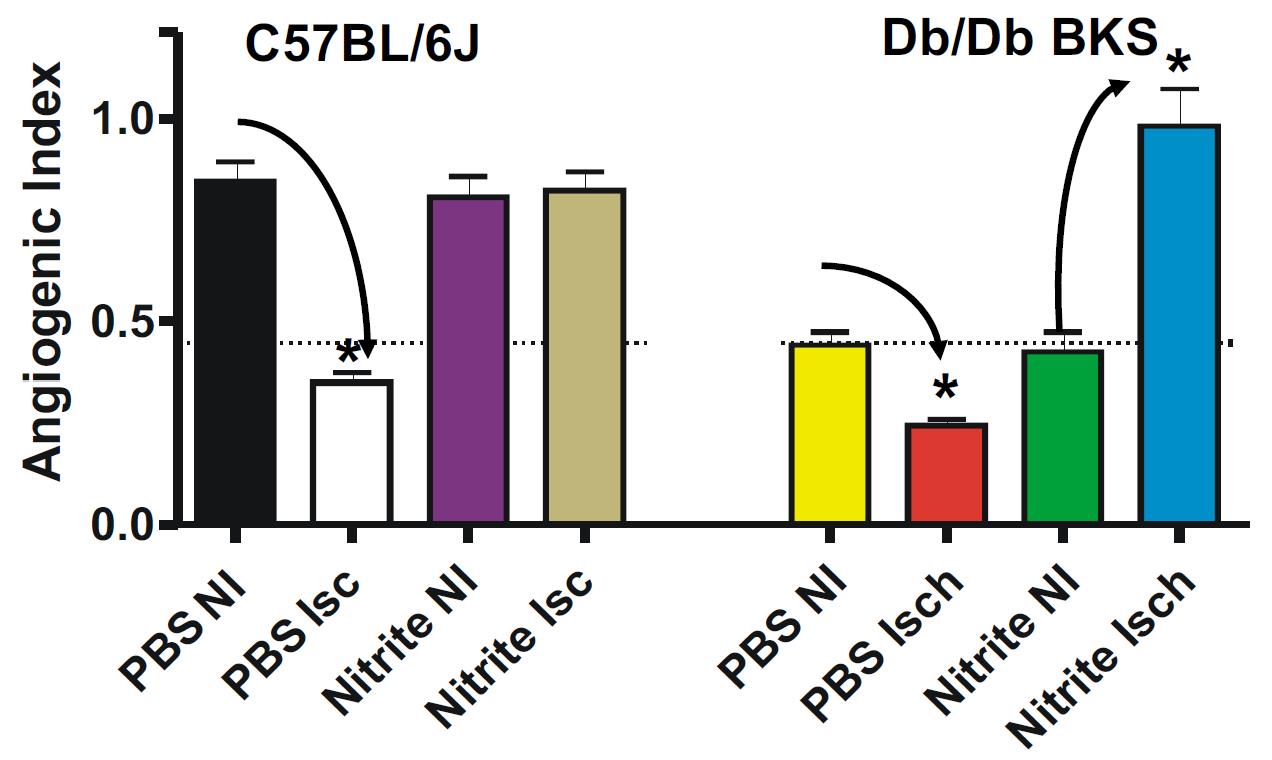
8
Nitrite therapy prevented ischemia mediated endothelial cell density loss in normal C57BL/6J ischemic limbs. Nitrite therapy significantly restored endothelial cell density in ischemic limbs of diabetic mice to normal C57BL/6J levels compared to PBS therapy of non-ischemic and ischemic conditions. These data suggest that nitrite therapy may be useful in attenuating microvascular rarefaction due to loss of nitric oxide that is observed during metabolic dysfunction (Frisbee JC AJP Integr Comp Physiol 2005 289(2):R307-16; Stepp et al Microcirculation 2007 14(4-5): 311-6).
Delayed Nitrite Therapy Restores Ischemic Hind-Limb Blood Flow
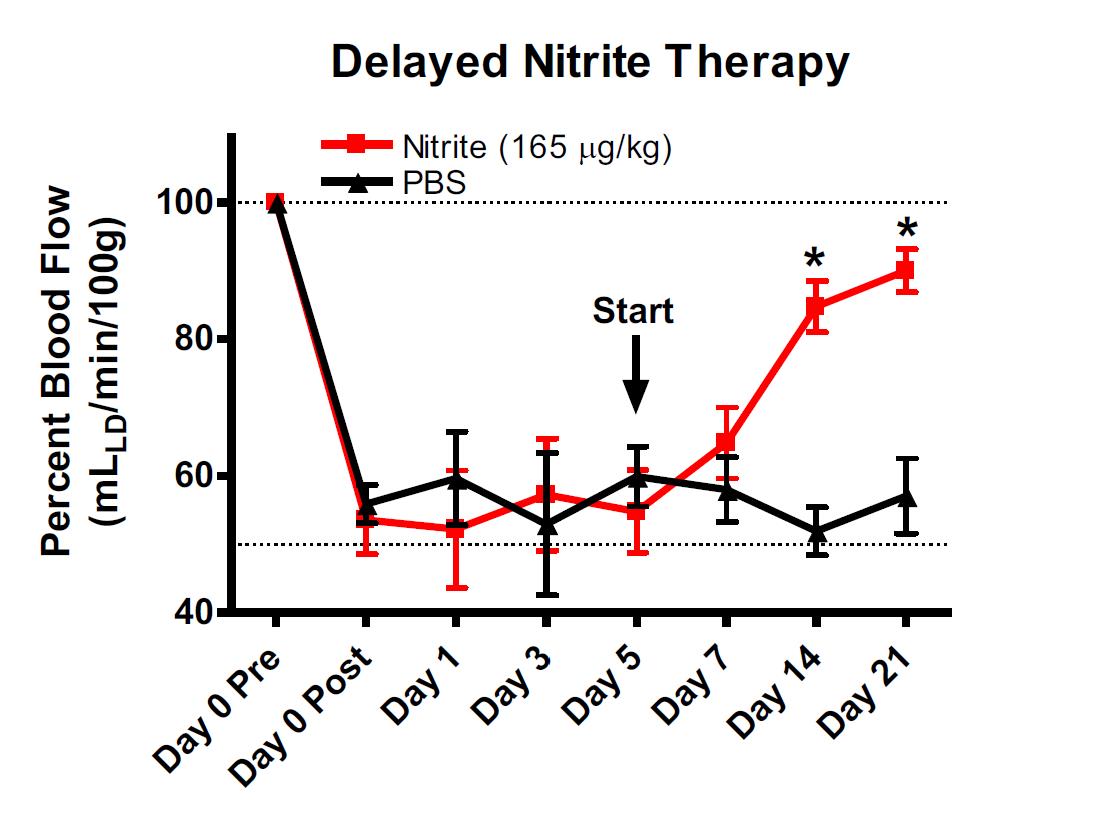
Studies were performed to determine whether nitrite mediated therapy would be effective in tissue that had been left ischemic for 5 days after femoral artery ligation. Femoral artery ligation was performed in C57BL/6J mice and the animals randomized to either PBS or sodium nitrite therapy 5 days after artery ligation. Treatments were given b.i.d. via I.P. injection. Ischemic limb blood flow was measured using laser doppler flowmetry. (Bir et al Diabetes 2014, 63(1):270-81).
9
Delayed nitrite therapy increases SPY angiogram arteriogenesis
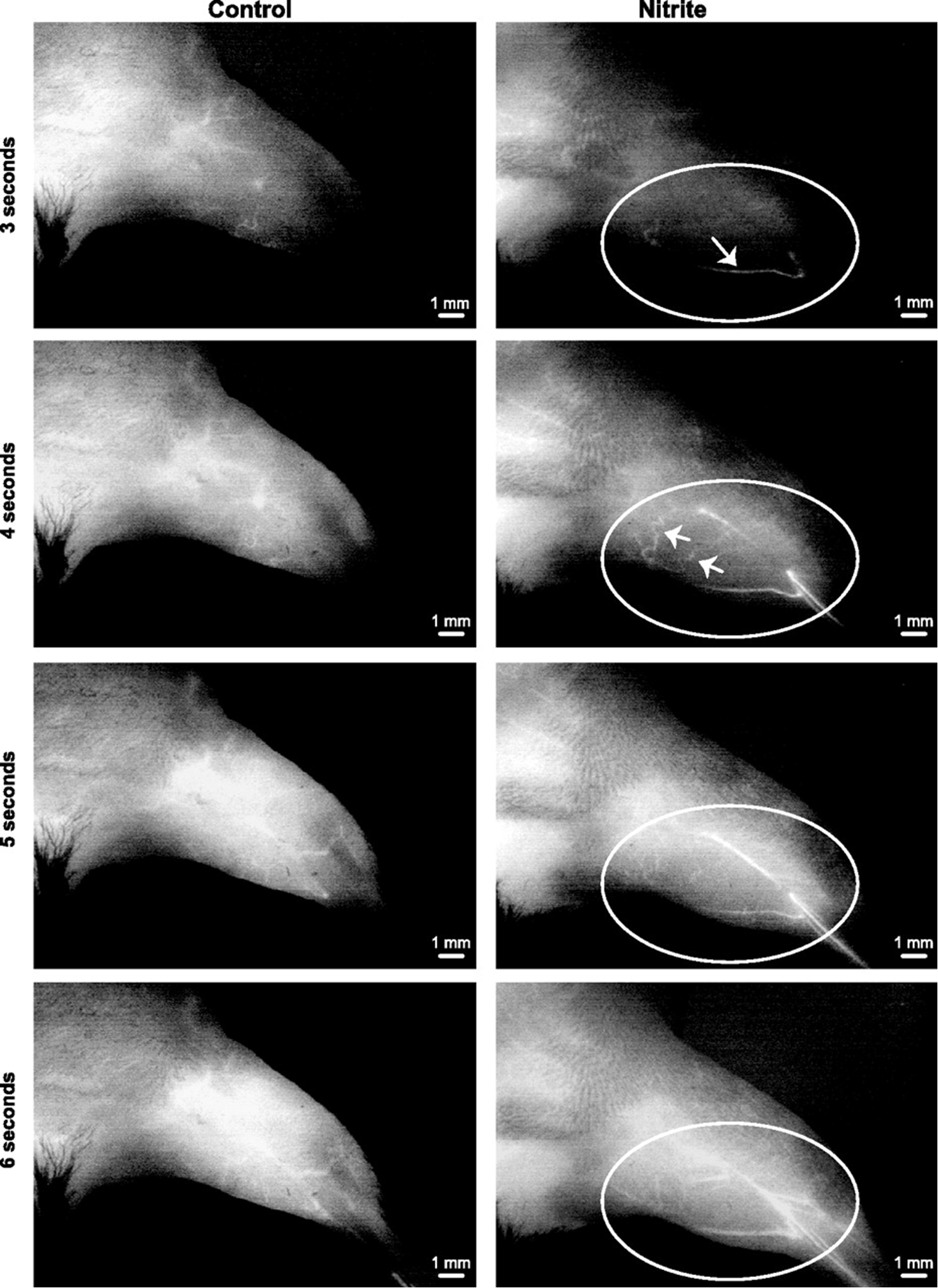
Delayed nitrite therapy increases SPY angiogram arteriogenesis. Representative temporal SPY angiogram image stills (3–6s) are shown at 11 days following ligation and 6 days after beginning therapy (either PBS or sodium nitrite). Left: PBS control angiogram. Right: sodium nitrite angiogram following injection of ICG. n = 5 animals per cohort. Circles identify limb anatomical regions of vascular blush, whereas arrows indicate perfused vessels that progressively occur over time.
Bir S C et al. Am J Physiol Heart Circ Physiol 2012;303:H178-H188.
10
Nitrite Therapy Prevents Tissue Necrosis in Aged Db/Db Mice
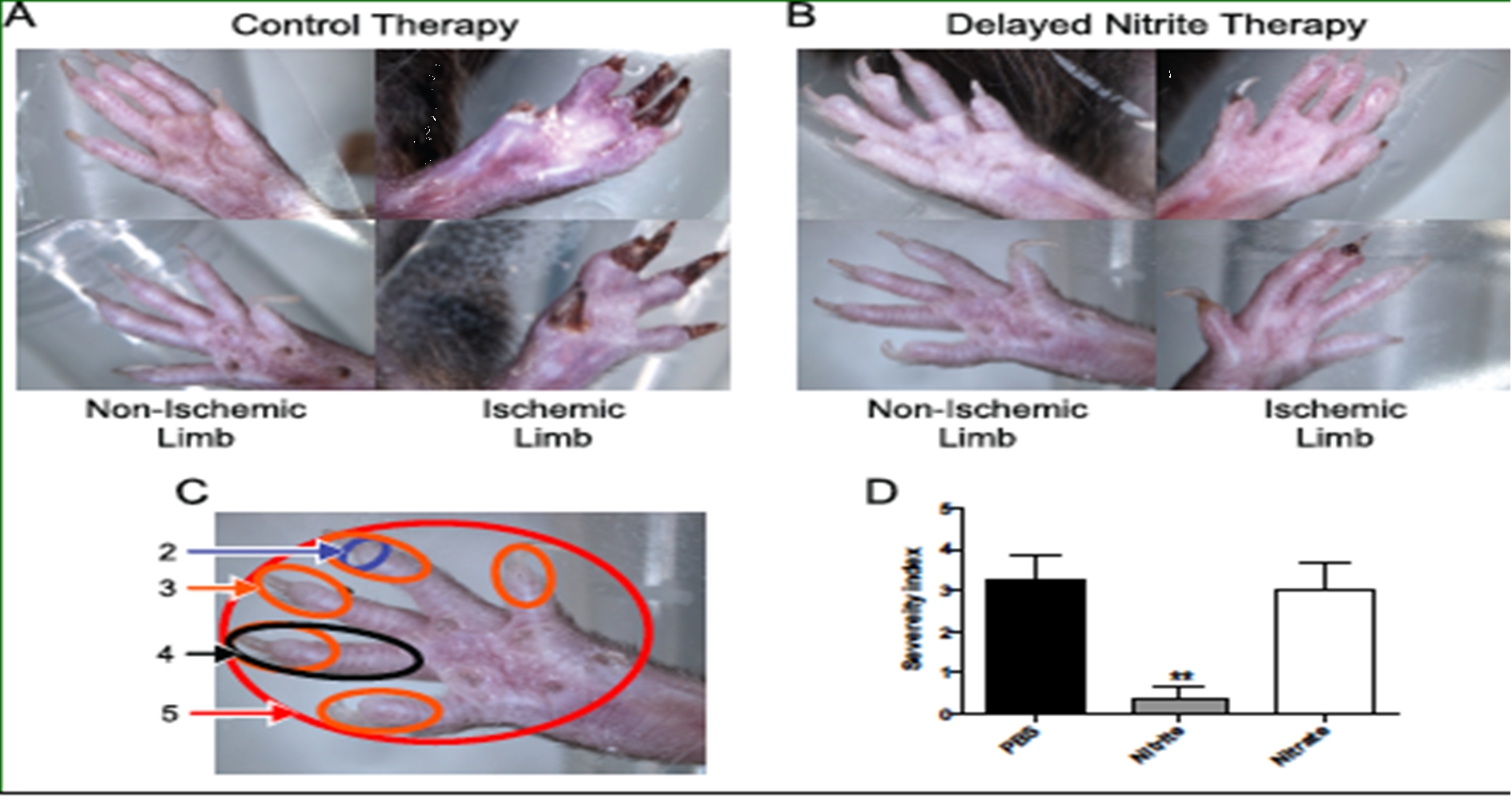
Delayed sodium nitrite (165 ug/kg) or control PBS therapy was stated 5 days post femoral artery ligation in 9 month old Db/Db mice. Nitrite therapy significantly prevented tissue necrosis (panel B) compared to control PBS therapy (panel A). Panel D reports tissue necrosis severity as a function of degree of limb and digit involvement. Nitrite therapy but not PBS control or sodium nitrate significantly prevented tissue necrosis. (Bir et al Diabetes 2014, 63(1):270-81).
Nitrite and Hind Limb Ischemia Summary
Sodium nitrite has long been known to be a potent vasodilator (transiently increasing blood vessel diameter) that can lead to a drop in blood pressure when given acutely. The above studies indicate that chronic administration at low doses, promotes angiogenesis, unlike single one-time nitrite therapy which does not stimulate angiogenesis. In addition, these studies and a large number of other studies not reviewed above, show:
|
|
• |
Nitrite therapy is very specific, acting only in damaged, ischemic tissue; |
|
|
• |
Delayed nitrite therapy effectively restores ischemic tissue blood flow; |
|
|
• |
Nitrite therapy is effective in a wide range of pathologies involving alterations of angiogenesis including critical limb ischemia, heart failure, and tissue necrosis; |
|
|
• |
Nitrite supplementation has had positive effects in various diabetes models, including diabetic nephropathy and diabetic wound healing; |
|
|
• |
Beneficial effects center on enhancing angiogenesis, endothelial cell proliferation, and arteriogenesis; and |
|
|
• |
Sustained release nitrite therapy, unlike immediate release therapy, does not lead to vasodilation or a drop in blood pressure. |
11
Our Product Candidate JAN101
Our product candidate is designed to treat diseases associated with poor vascular function. The following table summarizes our current product candidate pipeline:
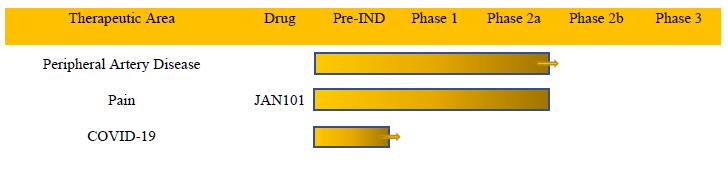
Therapeutic Area Peripheral Artery Disease Pain COVID-19 Drug JAN101 Pre-IND Phase 1 Phase 2a Phase 2b Phase
In March 2021, we determined to defer our current, on-going pursuit of JAN101 as a potential treatment for COVID-19 vascular complications. In connection with that determination, we will not currently file a utility patent nor submit an investigational new drug application to the FDA. This determination was based on several factors, including the current, positive effect that three vaccines are having on the COVID-19 pandemic in the United States. Should the vaccines ultimately prove less effective than currently expected or should new COVID-19 variants result in another pandemic, we may then determine to end our deferral of pursuing JAN101 as a treatment of COVID-19 vascular complications.
Pain
Pain is a protective reaction that alerts the body to the presence of actual or potential tissue damage so that necessary corrective responses can be mounted. The National Institutes of Health (the “NIH”) defines chronic pain as pain that persists beyond the normal healing time of an injury or that persists longer than three months. It is estimated that chronic pain affects 100 million individuals in the US and over 1.5 billion people worldwide, thus more people suffer from chronic pain than diabetes, heart disease and cancer combined (Cowen Therapeutic Categories Outlook March 2019). Chronic pain exacts a tremendous cost in terms of direct treatment and rehabilitation expenditures, lost worker productivity, prevalent addiction to opioid-based drugs, and emotional and financial burden for patients and their families. According to an Institute of Medicine of the National Academies report, pain is a significant public health problem in the United States that costs society between $560 billion and $635 billion annually. Despite the magnitude of the pain problem, innovation in the development of therapeutic solutions has been largely absent. Since 2010, there have been 20 approvals by the FDA for the treatment of pain, of which 12 were opioid variants, one was an extended release generic corticosteroid, five were variants of aspirin, and two were variants of other existing drugs. We are developing a novel product candidate designed to overcome the limitations of current treatment options for patients with PAD who suffer from chronic pain. According to a research study by Stanford University more than 24% of patients with PAD are at risk of high opioid use. By treating pain at the source and present patients and physicians with better and safer treatment alternatives we expect to minimize opioids at the prescription pad. Given the properties of JAN101, we have made the strategic decision to initially focus on pain associated with PAD by treating the underlying cause of PAD.
Peripheral artery disease
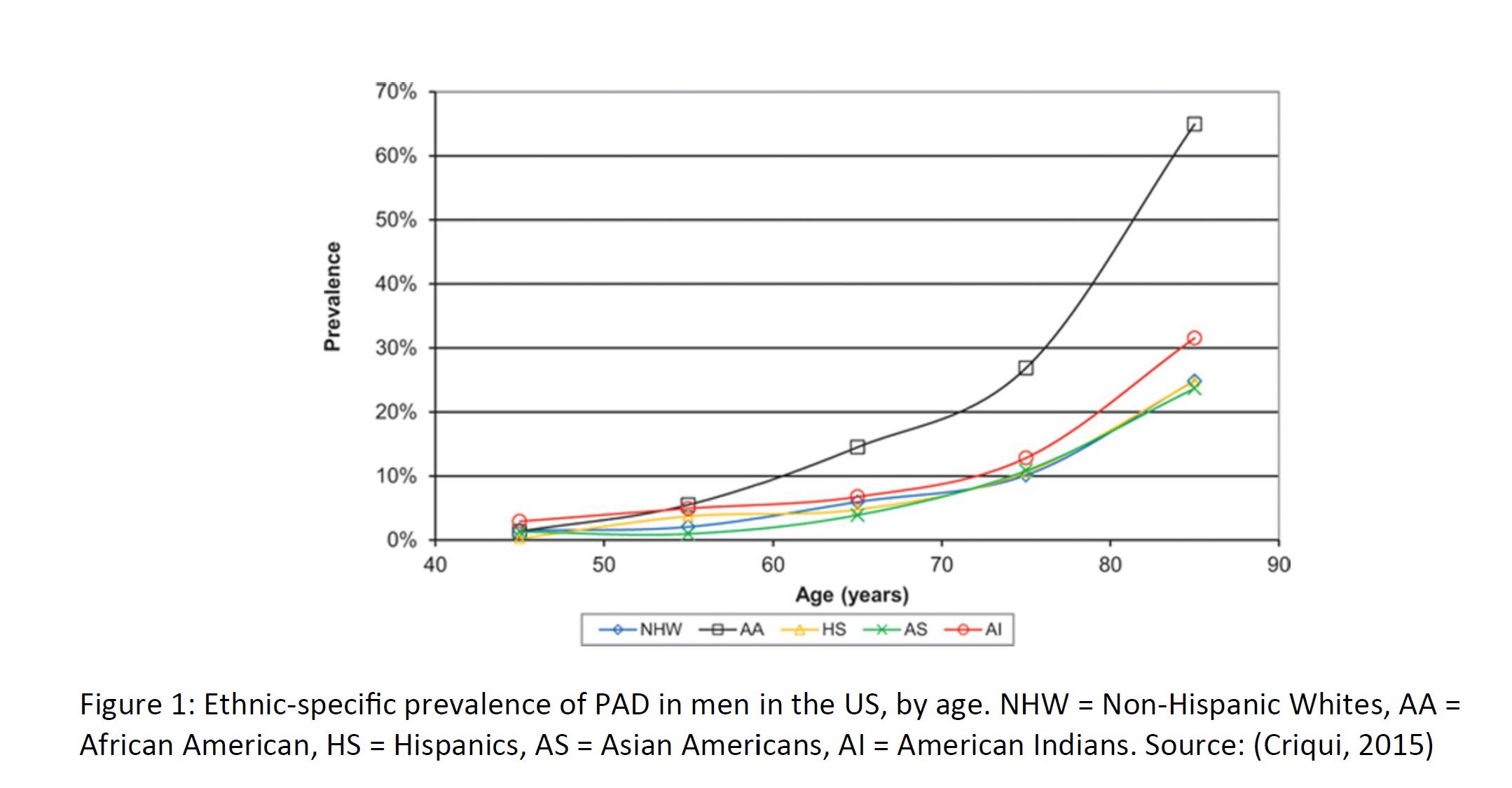
Peripheral artery disease is a general term for conditions in which arterial blood flow to the limbs are partially blocked. When there is less blood present in the extremities relative to demand, muscle pain and fatigue result, especially in the calf, which is also known as intermittent claudication. In many patients, pain and fatigue are relieved through rest. Roughly half of patients with PAD are asymptomatic. The most common cause of PAD / intermittent claudication is atherosclerosis. Diabetes, chronic kidney disease, hypertension, and smoking are all risk factors which can increase the likelihood of PAD. In atherosclerosis, fat deposits (plaques) build up along arterial walls, resulting in a reduction in blood flow in the legs. This same process can cause strokes if the arteries leading up to the brain are affected.
12
Because of the high rate of asymptomatic patients, prevalence figures vary widely. Some estimate that up to 200 million worldwide have PAD, ranging from asymptomatic disease to severe. Prevalence increases as a function of patient age, rising sharply after the age of 60. Thus, in countries with an aging population, it is expected that the prevalence of PAD will only increase. There is also a strong ethnic and racial component to PAD prevalence, which may be due to cultural differences in diet and exercise, along with genetic differences. Some suggest a prevalence of 8-12 million in the US alone, with roughly a third experiencing pain when walking, which improves upon resting. The diagnosis of PAD usually begins with patient complaints of pain in the extremities. If the patient is already being treated or monitored for diabetes or other risk factors, then the physician will check for a weak or absent pulse in the extremity. Decreased blood pressure, poor wound healing, and whooshing sounds in the legs (via stethoscope) are also tell-tale signs of PAD / intermittent claudication. Angiograms, electrocardiograms, and ultrasounds can also be used to image and confirm the diagnosis.
The non-drug treatment of PAD / intermittent claudication may be divided into four general categories:
|
|
• |
Lifestyle – Primarily changes in diet and smoking cessation. |
|
|
• |
Exercise – Patients who walk, cycle, stretch, or swim can experience marked improvement. Formal programs involving treadmills and track walking (usually 3-5 times per week) are frequently provided to patients. However, if the pain is triggered by exercise (claudication) and is significant, it can discourage the patient from exercise. |
|
|
• |
Angioplasty – A procedure by which the affected artery is stretched with a balloon-like device. This procedure has limited effectiveness and is reserved for severely blocked arteries. |
|
|
• |
Bypass Surgery – Arteries which are beyond angioplasty can be bypassed entirely. This procedure is typically reserved for cases where the blockage is considered very long (~10 centimeters) and nearly complete. |
The underlying condition, however, is not addressed by surgery. Surgical approaches will not, in the long run, improve exercise capacity and walking distance. Only exercise itself, coupled with lifestyle changes and drug approaches, has this benefit.
13
Prescription drugs for the treatment of the underlying PAD may be divided into multiple categories, depending on the underlying condition and severity:
|
|
• |
Cholesterol-Lowering Agents - Statins and bile acid sequestrants. |
|
|
• |
Antiplatelet Medica1ons – Aspirin and related drugs, such as clopidogrel. Cilostazol also has antiplatelet properties. |
|
|
• |
Antihypertensives – Patients with underlying high blood pressure can and will receive any number of medications to reduce blood pressure, such as ACE inhibitors and diuretics. |
|
|
• |
Diabetes Therapies – While a substantial portion of PAD patients may have pre-diabetes or fulminant diabetes, it is unknown if aggressive treatment of diabetes has a positive effect on PAD. |
|
|
• |
Pain – To our knowledge, no drugs are specifically indicated for PAD-associated pain. Pentoxifylline, for example, is indicated “…for the treatment of patients with intermittent claudication on the basis of chronic occlusive arterial disease of the limbs.” (Sanofi-Aventis U.S. LLC, 2010) However, the evidence supporting the effectiveness of pentoxifylline is mixed. Short-term courses of NSAIDs, such as ibuprofen may be used, provided the patient is not on another anticoagulant like aspirin. Non-drug pain relievers, such as TENS and massage therapy, may also be used in these patients. Opioids may also be used which creates a risk for addiction and potential misuse at the medicine cabinet by family members. |
The lack of any truly effective treatment of PAD, along with encouraging early trial results using JAN101 on both improving vascular function and reducing pain in PAD patients, has created an opportunity to potentially treat this large unmet medical need. By improving vascular function, JAN101 has the potential to reduce associated pain and improve PAD patients’ quality of life.
14
COVID-19
Coronavirus disease (COVID-19) is an infectious disease caused by a recently discovered coronavirus.
Most people infected with the COVID-19 virus will experience mild to moderate respiratory illness and recover without requiring special treatment. Older people, and those with underlying medical problems like cardiovascular disease, diabetes, chronic respiratory disease, and cancer are more likely to develop serious illness. The COVID-19 virus spreads primarily through droplets of saliva or discharge from the nose when an infected person coughs or sneezes.
One of the hallmarks of severe cases of COVID-19 is acute respiratory distress syndrome (“ARDS”), a rapid, widespread inflammation of the lungs that can lead to respiratory failure and death. In addition to the widely reported lung injuries associated with COVID-19, clinicians around the world are reporting that the disease also could be causing cardiac injuries in patients that sometimes lead to cardiac arrest. Kidney damage also is becoming a commonly reported issue among COVID-19 patients.
A study in the May, 28 2020 in the New England Journal published research detailing the post-mortem features of seven patients who died of COVID-19 provides critical insights, including evidence of extensive damage to the lining of the blood vessels, abnormal blood vessel growth in the lungs and widespread blood clotting. The study led by Steven Mentzer, HMS professor of surgery at Brigham and Women’s Hospital, and done in collaboration with a team of international researchers showed that infection with SARS-CoV-2, the virus that causes COVID-19, caused severe damage to the endothelial cells that line blood vessels and triggered widespread blood clotting. The team also identified signs of a distinctive pattern of vascular disease progression in some cases of COVID-19 compared with patterns seen in equally severe influenza virus infection. The findings highlight these key takeaways:
|
|
• |
While caused by a respiratory virus, COVID-19 manifests as a vascular disease that leads to severe injuries to blood vessels throughout the lungs. The damage to vascular cells may help explain why serious blood clotting has been observed in many patients. |
|
|
• |
The substantial new blood vessel growth seen in the lungs of COVID-19 patients occurs primarily through a mechanism known as intussusceptive angiogenesis—the splitting of existing blood vessels to form new ones—perhaps as a repair response to blood clotting and blood vessel damage, according to the authors. |
Damaged blood vessels may also underlie other problems, such as COVID toe, multisystem inflammatory syndrome in children (MIS-C), stroke and other seemingly unrelated problems seen with COVID-19.
In March 2021, we determined to defer our current, on-going pursuit of JAN101 as a potential treatment for COVID-19 vascular complications. In connection with that determination, we will not currently file a utility patent nor submit an investigational new drug application to the FDA. This determination was based on several factors, including the current, positive effect that three vaccines are having on the COVID-19 pandemic in the United States. Should the vaccines ultimately prove less effective than currently expected or should new COVID-19 variants result in another pandemic, we may then determine to end our deferral of pursuing JAN101 as a treatment of COVID-19 vascular complications.
Our Team
Tony Giordano PhD, our Chief Scientific Officer, joined the company in December 2019. Dr. Giordano joined JanOne from the Cleveland Clinic, the No.2 rated hospital in the country, where he served as Senior Director of Special Projects in the Business Development group. Dr. Giordano has extensive experience in commercialization and drug development, having served as Vice President or President of seven different biotechnology companies he co-founded, including companies developing platform technologies, a cancer vaccine, and Alzheimer’s Disease and cardiovascular therapies. He has managed numerous clinical trials and the launch of a medical food product. Dr. Giordano has also served as an Associate Professor and Assistant Dean of Research and Business Development at LSU Health Sciences Center in Shreveport, where he led the licensing efforts at the campus and at Abbott Labs, where in addition to serving as a Senior Research Scientist, he was involved in technology assessment activities. Dr. Giordano has a PhD focused in Molecular Genetics from Ohio State University and completed Fellowships at the NCI and NIA.
15
Dr. Amol Soin, our Chief Medical Officer, joined the Company in January 2020. Dr. Soin is considered one of the nation's top pain experts and is the Founder and Chairman of the Ohio Pain Clinic. Dr. Soin brings significant expertise for treating neuropathic and chronic pain and extensive research experience for non-opioid, nonaddictive pain solutions to the JanOne management team. In his role as Chief Medical Officer, Dr. Soin will guide JanOne's drug development activities, manage clinical research, set patient safety standards, and ensure regulatory compliance. In addition, Dr. Soin will play an integral role in establishing partnerships and drug candidate selection as the company expands its pipeline. Dr. Soin received his undergraduate degree from University of Akron, his MBA from University of Tennessee, his MD from Northeastern Ohio Universities College of Medicine, his master's in science from Brown University and also has studied at Dartmouth College. He is board certified in anesthesiology and pain medicine and a fellow of interventional pain management at the World Institute of Pain, and he served as a pain management fellow at the Cleveland Clinic, the oldest and largest academic pain management department in the United States. The founder and chairman of the Ohio Pain Clinic, Dr. Soin has also held several prestigious positions including President of the Ohio Society of Interventional Pain Physicians, president of the American Society of Interventional Pain Physicians Foundation, President of the Society of Interventional Pain Management Surgery Centers and president – elect of TriState Pain Society. He was appointed by Governor Kasich to the Ohio Medical Board in 2012 to two 5-year terms and has served as the Ohio Medical Board's president where he was instrumental in passing statewide rules and guidelines to help the opioid crisis.
In November 2019, we formed a Scientific Board of Advisors (the “SBA”) and the following doctors and scientist currently sit on the SBA:
Chris Kevil, Ph.D., Chair of the Scientific Advisory Board -- Dr. Kevil, an internationally known expert in vascular pathophysiology, PAD, and nitric oxide biology, discovered the role of sodium nitrite in promoting angiogenesis that led to the development of TV1001 now known as Jan101. Dr. Kevil earned his Ph.D. degree from LSU Health Shreveport in Molecular and Cellular Physiology followed by a fellowship at the University of Alabama at Birmingham (UAB) with an emphasis on redox pathophysiology. Returning to LSU Health Shreveport in the Department of Pathology, he established cutting edge research programs regarding redox biology regulation of peripheral vascular diseases. This led to ground-breaking insights on how glutathione, nitrite/nitric oxide, and hydrogen sulfide regulate vascular health during ischemia.
Edgar Ross, MD -- Dr. Ross is the current Director of the Pain Management Center at Brigham and Women's Hospital and a professor of anesthesia at Harvard Medical School. Dr. Ross is recognized as Castle Connolly's America's top doctors for the fifth year in a row. In addition to serving as chairman of Pfizer's partnership on pain, Dr. Ross also has served as a member of the Blue Cross and Blue Shield Opioid Prescribing Policy Committee.
Rakesh Patel, Ph.D. -- Dr. Patel is currently Vice Chair for Research, Department of Pathology, and Director of the Center for Free Radical Biology at the University of Alabama at Birmingham (UAB). Most noted is his research to understand the molecular basis of nitric oxide, and nitrite interactions with organs and red blood cells. Patel is also known for his work to understand the impacts on the biological process associated with blood flow regulation and pulmonary function.
Timothy Ness, MD, Ph.D. -- Dr. Ness is Professor Emeritus and former Pain Treatment Division Chief, Director of Pain Research and Vice Chair for Clinical Research in the Department of Anesthesiology and Perioperative Medicine at the University of Alabama at Birmingham (UAB) He has served as a clinical research expert on pain for the National Institutes of Health (NIH), Food and Drug Administration (FDA) advisory panels, the Veterans Administration (VA), and various international research institutes. He has served on the American Pain Society and the American Society of Regional Anesthesia and Pain Medicine Board of Directors. He is currently funded by the NIH.
Alan Kaye, MD, PhD, DABA, DABPM, DABIPP -- Dr. Kaye is the Professor and Chairman of the Department of Anesthesiology at LSU Health Sciences Center in New Orleans. Before LSU, he was Professor and Chairman of the Texas Tech University Health Sciences Center Department of Anesthesiology in Lubbock, Texas. Prior, he was the Medical Director of the Greater New Orleans Surgical Center, the Director of Resident Recruitment, Acting Program Director and an Attending Staff of the Department of Anesthesiology at Tulane University Medical Center in New Orleans. He received two BS degrees and a MD degree from the University of Arizona. He also completed a pain management fellowship at Texas Tech Health Sciences Center. He is Board Certified as a Consultant in
16
Anesthesiology and has a special certificate in Pain Management for the American Board of Anesthesiology. He is also a Diplomate of the American Board of Pain Medicine and the American Board of Interventional Pain Physicians. Dr. Kaye completed his PhD in pharmacology in May 1997. His thesis title was "Pharmacology of Angiotensin Peptides and Nonpeptide Agonists in the Pulmonary Vascular Bed of the Cat and of the Rat." He authored or co-authored over 150 abstracts and 200 manuscripts and book chapters in the fields of pulmonary vascular pharmacology and anesthesiology. He serves on a number of national committees including as a National Board of Directors of ASIPP and ABIPP. He is editor-in-chief of the journal Pain Physicians and is on the FDA Advisory Board on Anesthetics and Analgesics. He was an Associate National Board Examiner in Anesthesiology
John Cooke, MD, Ph.D. -- is the Chair of the Department of Cardiovascular Sciences at the Houston Methodist Research Institute, Director of the Center for Cardiovascular Regeneration, and Medical Director of the RNA Therapeutics Program in the Houston Methodist DeBakey Heart and Vascular Center in Houston, Texas. He trained in cardiovascular medicine and obtained a Ph.D. in physiology at the Mayo Clinic. He was recruited to Harvard Medical School as an assistant professor of medicine. In 1990, he was recruited to Stanford University to spearhead the program in vascular biology and medicine, and was appointed professor in the Division of Cardiovascular Medicine at Stanford University School of Medicine, and associate director of the Stanford Cardiovascular Institute until his recruitment to Houston Methodist in 2013. Dr. Cooke has published over 500 research papers, position papers, reviews, book chapters and patents in the arena of vascular medicine and biology with over 30,000 citations. He has served on national and international committees that deal with cardiovascular diseases, including the American Heart Association, American College of Cardiology, Society for Vascular Medicine, and the National Heart, Lung and Blood Institute. He has served as president of the Society for Vascular Medicine, as a director of the American Board of Vascular Medicine, and as an associate editor of Vascular Medicine.
Our Strategy
Our mission is to develop and commercialize novel, non-opioid, and non-addictive therapies to safely and effectively address the significant unmet medical need of chronic pain or treat conditions that cause pain. The principal elements of our strategy to achieve this mission are the following:
|
|
• |
License, acquire, develop, and create novel, non-opioid and non-addictive therapies by leveraging our understanding of pain biology to address the large and growing problem of pain. While innovation in medical sciences has led to exciting new treatment options in many disease areas, pain has seen limited innovation in recent years. We have a deep understanding of the pathophysiology of pain and diseases that cause pain. We intend to leverage this understanding to bring innovation in the pain treatment paradigm through targeted acquisitions of companies or assets in development. Our advisors and doctors have years of collective experience in leadership positions at institutions and substantial scientific experience and understand the complexity of designing and executing clinical trials for and developing therapies. |
|
|
• |
Advance the development of our lead product candidate, JAN101, designed for the treatment of patients with PAD and pain associated with the disease. There are limited therapeutic options available for patients with PAD and we believe that JAN101 has the potential to transform the standard of care to a twice a day pill to substantially improve moderate to severe PAD. The company plans to engage a contract research organization (“CRO”) in early 2021 and begin enrolling subjects for the first Phase 2b trials for JAN101, and we expect to report topline results promptly following receipt of the data from the CRO. |
|
|
• |
Leverage clinical activity of JAN101 to possibly expand into new indications, including complications associated with COVID-19We believe that JAN101 may have utility in treating vascular complications in patients with COVID-19 as we believe COVID-19 is an endothelial cell disease which manifests its complications in the vascular system and major organ causing complications in recovered patients. In November 2020, we filed an investigational new drug application for our COVID-19 indication (which was subsequently converted to a pre-IND). In March 2021, we determined to defer our current, on-going pursuit of JAN101 as a potential treatment for COVID-19 vascular complications. In connection with that determination, we will not currently file a utility patent nor submit an investigational new drug application to the FDA. This determination was based on several factors, including the current, positive effect that three vaccines are having on the COVID-19 pandemic in the United States. Should the vaccines ultimately prove less effective than currently expected or should new COVID-19 variants result in another pandemic, we may then determine to end our deferral of pursuing JAN101 as a treatment of COVID-19 vascular complications. |
17
|
|
• |
Advance our product candidates through clinical development and pursue development of additional product candidates through acquisitions. Our objective is to build a well-balanced, multi-asset portfolio targeting the large population of patients with chronic and acute pain. To achieve this, in addition to JAN101, we intend to pursue partnerships, licensing agreements, and potential acquisitions of other pharma companies. We continue our search for assets with indications where we believe they could have meaningful impact and address the large unmet medical need. In addition, we may choose to selectively in-license or acquire complementary product candidates by leveraging the insights, network, and experience of our team. |
|
|
• |
Maximize the commercial potential of all our product candidates. We currently intend to retain all commercial rights to JAN101 in the United States and selectively partner outside of the United States. Because we believe that PAD is an attractive market for many major pharmaceutical companies, we may sub-license or partner certain indications if we believe it may enhance stockholder value. As we continue to build and develop our product portfolio, we may opportunistically pursue strategic partnerships that maximize the value of our pipeline while seeking to develop other indications. |
|
|
• |
Leverage our management team background and expertise. We have assembled a team with extensive experience described above. |
Chronic Pain
The NIH defines chronic pain as pain that persists either beyond the normal healing time of an injury or longer than three months. We believe that chronic pain represents a significant public health crisis. In the United States, chronic pain affects approximately 40 million adults annually, which is greater than the annual prevalence of each of heart disease, cancer and diabetes. It is also estimated that pain leads to between $560 and $635 billion in healthcare and lost productivity costs each year. Chronic pain is the leading cause of long-term disability in the United States, and approximately 23 million adults in the United States experience severe pain over a three-month period. Globally the prevalence of chronic pain is even larger, with over 1 billion people worldwide affected each year. Common types of chronic pain include those of neuropathic and inflammatory origin and may involve the skin, muscles, joints, bones, tendons, ligaments, and other soft tissues. Chronic pain is associated with a variety of clinical conditions including, but not limited to, arthritis, spinal conditions, cancer, fibromyalgia, diabetes, surgical recovery, visceral injury and general trauma.
Pain is a necessary protective reaction that alerts the body to the presence of actual or potential tissue damage so that necessary corrective responses can be mounted. Pain is signaled by specialized cells in the peripheral nervous system called nociceptors, or pain-sensing fibers. These pain-sensing fibers normally transmit information about stimuli that approach or exceed harmful intensity from different locations in the body to the brain, which registers this information as a sensation of pain. In the case of tissue injury due to trauma or infection, pain accompanies the associated inflammation, persists for the duration of the inflammatory response, and aids healing by inhibiting use of the affected body part.
Pain also can modify the central nervous system such that the brain becomes sensitized and registers more pain with less provocation. This is called central sensitization. When central sensitization occurs, the nervous system goes through a process called wind-up and gets regulated in a persistent state of high reactivity. This persistent, or up-regulated, state of reactivity lowers the threshold for what triggers the sensation of pain and can result in the sensation of pain even after the initial injury might have healed.
When there is dysfunction in pain signaling, injury to the nervous system, or an unhealed injury, pain becomes no longer just a symptom, but a disease in itself.
Current Therapeutic Approaches to Treating Chronic Pain and Their Limitations
NSAIDs
Some of the most widely used therapies to treat chronic inflammatory pain are non-steroidal anti-inflammatory drugs, or NSAIDs. NSAIDs can have significant side effects that include gastrointestinal bleeding, gastritis, high blood pressure, fluid retention, kidney problems, heart problems and rashes. On April 7, 2005, the FDA announced a decision to require boxed warnings of potential cardiovascular risk for all NSAIDs.
18
Corticosteroids
Corticosteroids, or steroids, also possess anti-inflammatory properties and are commonly used in the practice of pain management, either systemically or locally, depending on the condition. Steroids work by decreasing inflammation and reducing the activity of the immune system. While steroids are commonly used, they may have numerous and serious side effects. These side effects may include allergic or hypersensitivity reactions, increased risk for infection, adrenal insufficiency, diabetes or decreased glucose tolerance, hypertension, loss of bone density, and loss of joint cartilage volume. In addition, steroids should not be administered when there is an infection present because steroids can inhibit the body’s natural infection-fighting immune response. Also, if a joint is already damaged or is subject to chronic deterioration, intra-articular, or IA steroid injections are not likely to provide any long-term restorative benefit. For the above reasons, IA steroid injections are generally recommended to be administered no more often than every six weeks and not more than three to four times per year.
Opioids
Opioids are some of the most widely prescribed therapeutics for chronic and acute pain, and sales of these drugs have quadrupled between 1999 and 2010. According to a National Survey on Drug Use and Health report, in 2016 more than one third of adult Americans were prescribed opioids and 230 million opioid prescriptions were written that year in the United States. Opioids act by binding to specific receptors located on neurons in both the central and peripheral nervous system throughout the body including in the brain, spinal cord and other nervous tissue. Although they can be effective in providing pain relief, the increased medical use of opioids has been accompanied by an increase in the abuse and misuse of prescription opioids. In addition, for most patients, chronic opioid use is a poor option due to an intolerance to the many side effects, including nausea, vomiting, drowsiness and constipation, and the propensity for opioids to become less effective with long-term use. According to the Centers for Disease Control and Prevention, or CDC, almost two million individuals abused or were dependent on prescription opioids in 2014. CDC figures show that the number of opioid-related overdose deaths has quadrupled between 1999 and 2010, and currently approximately 40% of opioid overdose deaths in the United States involve a prescription opioid. This increase in prescription opioid-related deaths in the United States prompted former President Trump to declare the opioid crisis a national Public Health Emergency in October 2017. Opioid abuse has become an epidemic in the United States, ranking as the nation’s second most prevalent illegal drug problem. These major issues create the need to find new approaches to treating chronic pain.
Our Approach to Treating PAD and Chronic Pain
The unmet medical need for treating PAD and chronic pain reflects the historic failure to develop novel classes of analgesics with comparable or greater efficacy, an acceptable level of adverse effects and a lower abuse liability than those currently available. Some of the reasons for this include the heterogeneity of chronic pain and its related conditions, and the complexity and diversity of the underlying pathophysiological mechanisms for pain. However, recent advances in the understanding of the neurobiology of pain are beginning to offer opportunities to identify new drug targets and develop new therapeutic strategies.
We have taken an innovative and targeted approach to identifying treatments for chronic pain that leverages our understanding of the pathophysiology of pain. Pain is variable—for example, it can be inflammatory or neuropathic in nature, and it may be localized to a specific area of the body or it may be generalized throughout. We believe that the most effective way to treat chronic pain is through therapies that specifically target the origin of the pain signal. We strive to maximize each of our product candidate’s potential based on its unique mechanism of action related to the origin of the pain signal.
19
A Randomized, Double-Blind Study of the Effects of a Sustained Release Formulation of Sodium Nitrite (SR-nitrite) on Patients with Diabetic Neuropathy
Background: Background: Sodium nitrite has been reported to be effective in reducing chronic peripheral pain.
Objectives: To evaluate the safety and efficacy of 40 and 80 mg, BID, of an oral sustained release formulation of sodium nitrite (SR-nitrite) in patients suffering from diabetic neuropathy, and to determine whether SR-nitrite would reduce the frequency of headaches reported previously by subjects receiving the same doses of an immediate release formulation. Study Design: Phase II, single-center, randomized, double-blind, placebo controlled clinical trial. Setting: The Ohio Pain Clinic and Kettering Medical Center.
Methods: Twenty-four patients were randomized to 40 mg or 80 mg SR-nitrite or placebo twice daily for 12 weeks. The primary objective was to determine whether headaches would be reduced using SR-nitrite. The primary efficacy endpoint was the mean difference in the change of the Neuropathic Pain Symptom Inventory (NPSI) pain score from baseline to that reported after 12 weeks of treatment. Secondary endpoints included changes from baseline for the Brief Pain Inventory (BPI) Scale, the RAND 36 questionnaire, Short Form McGill Questionnaire, daily patient reported score for neuropathic pain, changes in HbA1c, PulseOx and quantitative sensory testing. Results: The number of subjects reporting adverse events and the number of adverse events did not change with dose. There were no reports of treatment-related headaches. Although no significant differences were identified in patient responses to the questionnaires, a trend was observed. In the NPSI assessment, patients in the 40 mg and 80 mg dose group reported a 12.7% and 22.0% reduction in pain, respectively, compared to an 8.4% reduction by patients in the placebo group. A trend was also observed with the BPI total severity score. However, the 40 mg dosing group reported the greatest reduction in pain using the McGill Pain index and via patient logs of daily pain scores, where the mean of pain scores reported by subjects in the 40 mg group dropped by day 41 and generally stayed lower than the mean of scores reported by subjects in either of the other two groups. Patients in the 80 mg SR-nitrite group had an improvement in both Nerve Sensory Conductance and Nerve Sensory Velocity. No changes were observed in HbA1c levels or PulseOx.
Limitations: Small sample size.
Conclusion: Sustained release sodium nitrite prevents the prevalent reports of headaches by patients treated with an immediate release formulation of sodium nitrite. In a previous study of patients with peripheral arterial disease (PAD), 40 mg BID treatment led to a statistically significant reduction in reported pain, similar trends were observed at the end of the trial period for most of the pain questionnaires used in the study. The 80 mg BID treatment had the more pronounced affect on bioactivity (quantitative sensory testing), which was similar to the PAD study, where this dose group had the greatest improvement in FMD {AU: spell out FMD}. The ability to alleviate pain with BID treatment of SR-nitrite offers promise for a new non-addictive, non-sedating treatment of chronic pain and warrants further study.
Microcirculatory injury, which is common in diabetic patients, can lead to a number of problems. Prominent among these is diabetic peripheral neuropathy (DPN) (1,2). About 10% of patients will have evidence of DPN at the time they are initially evaluated, and almost 50% of diabetic patients will ultimately develop DPN. Of diabetic patients with DPN, 40% to 50% suffer from chronic pain as well as paresthesias, sensory loss, and weakness, and have at least an 8-fold increased risk of undergoing a distal lower extremity amputation compared to similar non-diabetics. Endothelial cells play an important part in the regulation of microcirculation, as they maintain vascular tone by secreting both vasodilators and vasoconstrictors. A central feature of diabetic microvascular disease (MVD) is endothelial dysfunction, which, in turn, plays an important role in the development and progression of DPN. The pathophysiological factors leading to endothelial dysfunction in diabetes include chronic hyperglycemia and protein glycolation, insulin resistance, inflammation, and increased oxidative stress. Studies have now shown a close relationship between endothelial dysfunction and diminished nitric oxide (NO) bioavailability. Endogenously produced NO has a half life measured in seconds, and is rapidly oxidized to nitrite (NO2–) and nitrate (NO3––) end products, the latter of which is biologically inert. In the presence of microcirculatory ischemia and endothelial cell dysfunction, however, endogenous NO production by eNOS is much more limited. In such circumstances, circulating NO2– can be non-enzymatically reduced to increase NO availability. In addition to serving as a circulating NO reservoir, nitrite itself has also been shown to have direct and potent vasodilatory effects in vitro and in vivo. The findings that NO2– mediates vasodilatation, both directly and through NO generation, has led to
20
growing interest in the potential effectiveness of nitrite as a therapeutic agent in conditions associated with DPN and endothelial dysfunction. Such conditions include diabetic microvascular disease, DPN, and retinopathy, in which low levels of NO and NO2–, as well as elevated levels of nitrate (NO3), suggest that the complete oxidation of NO occurs during diabetes with insufficient NO2– reserves to restore NO bioavailability. Previous human studies with an oral formulation of NaNO2 have shown that administration twice daily improves vascular function. In the peripheral arterial disease study, subjects who received the lower dose of NaNO2 reported a significant reduction in pain. Although side effects were minimal, headaches and dizziness were reported by a large number of subjects, likely due to the rapid release of NaNO2 leading to vasodilation. An oral sustained-release formulation of NaNO2 (SR-nitrite) was developed in an attempt to overcome these problems and was tested in a porcine model of metabolic syndrome with critical limb ischemia. SR-nitrite-treated animals showed increased myocardial NO bioavailability, diminished oxidative stress, and cytoprotection in ischemic tissue. Importantly, 24-telometry recordings of blood pressure showed no evidence of vasodilation. In the above study, we hypothesized that the SRnitrite would reduce or eliminate headaches reported in patients following administration of the immediate release formulation. Given the promising results on reducing pain in diabetic patients with peripheral arterial disease reported in the previous study, patients with diabetic neuropathy were utilized in this study to determine whether any trends in reducing pain could be observed. The study design was a randomized, placebo controlled, double-blind phase II study was carried out to investigate the safety and potential biological activity of multiple doses of an oral, sustained-release formulation of sodium nitrite (SR-nitrite; Theravasc Inc., Cleveland, OH, USA), BID in doses of 40 mg and 80 mg over a 12-week treatment period, in human subjects with diabetes and neuropathic pain in the lower extremities and feet. The trial was approved by the Copernicus institutional review board and listed on ClinicalTrials.gov: www.clinicaltrials.gov/ct2/show/NCT02412852. The study was funded by Theravasc Inc.
JAN101—Regulatory Strategy
Sodium Nitrite has been previously approved as one of the active components of cyanide poisoning antidote. This means the approval path for JAN101 is through a 505(b)(2) new drug application (“NDA”), which we intend to pursue.
JAN101—Commercial Strategy
We currently intend to use third party providers and manufacturers to effectively support the commercialization JAN101, if we are successful in obtaining FDA approval. We believe that we can cost effectively promote JAN101 to the patients suffering from PAD. We anticipate our commercial operation to include outside sales management, outside sales support, distribution support and an internal marketing group. Additional requisite capabilities will include focused management of key accounts, such as managed care organizations, group purchasing organizations, and government accounts. We intend to selectively partner with third parties with vast experience in the space as we have been partnering for every aspect of development.
Competition
The biotechnology and pharmaceutical industries are characterized by extensive research and development efforts, rapidly advancing technologies, intense competition, and a strong emphasis on proprietary products. We are currently focused on the development and commercialization of our asset pipeline of novel, non-opioid and non-addictive therapies for PAD. The number of patients suffering from chronic PAD is large and growing. While we believe that our product candidate and our Chief Scientific Officers development experience and scientific knowledge provide us with competitive advantages, we face potential competition from many different sources, including pharmaceutical, biotechnology, and specialty pharmaceutical companies either marketing or developing therapeutics to treat chronic pain. Academic research institutions, governmental agencies, as well as public and private institutions are also potential sources of competitive products and technologies. Our competitors may have significantly greater financial resources, robust drug pipelines, established presence in the market and expertise in research and development, manufacturing, pre-clinical and clinical testing, obtaining regulatory approvals and reimbursement and marketing approved products than we do. These competitors also compete with us in recruiting and retaining qualified clinical, regulatory, scientific, sales, marketing and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. The key competitive factors affecting the success of all of our product candidates, if approved, are likely to be their efficacy, durability, safety, price and the availability of reimbursement from government and other third-party payors.
21
Significant competition exists in the PAD pain field. Although we believe our approach to developing novel treatments for pain is unique from most other existing or investigational therapies, such as NSAIDs, corticosteroids and opioids, we will need to compete with all currently available and future therapies within the indications where our development is focused. With respect to JAN101, the main classes of marketed products that are available for the treatment of PAD pain include NSAIDs and opioids. Furthermore, numerous monoclonal antibodies targeting nerve growth factor, or NGF inhibitors, are in clinical development, including two product candidates in Phase 3.
There are a number of companies developing or marketing therapies for the treatment and management of pain that may compete with our current product candidate, including many major pharmaceutical and biotechnology companies.
Intellectual Property
Our success depends in large part upon our ability to obtain and maintain proprietary protection for our products and technologies, and to operate without infringing or otherwise violating the proprietary rights of others. We endeavor to protect our products using a combination of intellectual property protections and available government regulatory and marketing exclusivities afforded to new medicines. For example, we endeavor to protect our products by, among other methods, filing U.S., and potentially in the future, foreign, patent applications related to our proprietary technology, inventions and improvements that are important to the development and implementation of our business. We also use other forms of protection, such as confidential information, trade secrets and know-how, and trademarks to protect our intellectual property, particularly where we do not believe patent protection is appropriate or obtainable.
The proprietary nature of, and protection for, our product candidates, processes and know-how are important to our business. Our policy is to pursue, maintain and defend intellectual property rights, and to protect the technology, inventions, and improvements that are commercially important to our business.
Trade Secrets and Other Proprietary Information
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. For example, we have developed methods for the more efficient manufacture of sustained released sodium nitrite tablets. We seek to protect our proprietary information, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and commercial partners.
License Agreement
On November 19, 2019, we entered into a Patent and Know How License Agreement (the “License Agreement”) with UAB Research Foundation (“UABRF”), TheraVasc, Inc. (“TheraVasc”), and the Board of Supervisors of Louisiana State University and Agricultural and Mechanical College, acting on behalf of LSU Health Sciences Center at Shreveport (“LSU Health Shreveport”, together with UABRF and TheraVasc, the “Licensors”). Under the License Agreement, the Licensors have agreed to grant to JanOne an exclusive, worldwide license, including the right to sublicense, to the Licensors’ patent rights and know-how related to the Licensors’ sustained release formulation of sodium nitrite. Under the License Agreement, we have agreed to pay a non-refundable upfront license fee and certain milestone payments upon the achievement of certain milestones of up to approximately $6.5 million and certain royalty payments and annual license maintenance fees. The License Agreement requires us to use commercially reasonable efforts to develop and commercialize JAN101.
22
Commercial Operations
We currently have no marketing and sales organization. We have retained global rights to our product candidate, and, if one of our product candidates is approved by the FDA to market in the United States we expect that our sales force will be supported by sales management, internal sales support, an outside marketing group and distribution support. We intend to invest in our commercial capabilities prudently by focusing our marketing efforts on the physician subspecialties that treat patients with PAD. These physicians include, but are not limited to, pain management specialists, rheumatologist, surgeons and sports medicine physicians. We will also evaluate licensing and partnering with third parties to help us reach other sales channels and geographic markets inside and outside of the United States.
Government Regulation
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries impose substantial and burdensome requirements upon companies involved in the clinical development, manufacture, marketing and distribution of drugs, such as those we are developing. These agencies, and other federal, state and local entities regulate, among other things, the research and development, testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, distribution, post-approval monitoring and reporting, sampling and export and import of our product candidates.
U.S. Government Regulation of Drug Products
In the United States, the FDA regulates drugs under the FDCA and its implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending applications, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
|
|
• |
completion of pre-clinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s good laboratory practice, or GLP, regulations; |
|
|
• |
submission to the FDA of an IND which must become effective before human clinical trials may begin; |
|
|
• |
approval by an institutional research board, or IBR, at each clinical site before each trial may be initiated; |
|
|
• |
performance of adequate and well-controlled human clinical trials in accordance with good clinical practice, or GCP requirements to establish the safety and efficacy of the proposed drug product for each indication; |
|
|
• |
submission to the FDA of an NDA; |
|
|
• |
satisfactory completion of an FDA advisory committee review, if applicable; |
|
|
• |
satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practices, or cGMP requirements and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
23
|
|
• |
satisfactory completion of FDA audits of clinical trial sites to assure compliance with GCPs and the integrity of the clinical data; |
|
|
• |
payment of user fees and securing FDA and approval of the NDA; and |
|
|
• |
compliance with any post-approval requirements, including the potential requirement to implement a risk evaluation and mitigation strategy, or REMS, and the potential requirement to conduct post-approval studies. |
Pre-clinical Studies
Pre-clinical studies include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess potential safety and efficacy. An IND sponsor must submit the results of the pre-clinical tests, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Some pre-clinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence. Clinical holds also may be imposed by the FDA at any time before or during clinical trials, due to safety concerns about on-going or proposed clinical trials, or non-compliance with specific FDA requirements, and the trials may not begin or continue until the FDA notifies the sponsor that the hold has been lifted.
Clinical Trials
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health for public dissemination on their www.clinicaltrials.gov website. The information contained in, or accessible through, this website does not constitute a part of this prospectus. We have included this website address in this prospectus solely as an inactive textual reference.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
|
|
• |
Phase 1: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness. |
|
|
• |
Phase 2: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
|
|
• |
Phase 3: The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product. |
Post-approval trials, sometimes referred to as Phase 4 trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA.
24
The FDA or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. In addition, some clinical trials are overseen by an independent group of qualified experts organized by the sponsor, known as a data safety monitoring board or committee. Depending on its charter, this group may determine whether a trial may move forward at designated check points based on access to certain data from the trial.
During the development of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase 2, and before an NDA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and the FDA to reach agreement on the next phase of development. Sponsors typically use the meetings at the end of the Phase 2 trial to discuss Phase 2 clinical results and present plans for the pivotal Phase 3 clinical trials that they believe will support approval of the new drug.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final drug. In addition, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
While the IND is active and before approval, progress reports summarizing the results of the clinical trials and non-clinical studies performed since the last progress report must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and investigators for serious and unexpected suspected adverse events, findings from other studies suggesting a significant risk to humans exposed to the same or similar drugs, findings from animal or in vitro testing suggesting a significant risk to humans, and any clinically important increased incidence of a serious suspected adverse reaction compared to that listed in the protocol or investigator brochure.
United States Review and Approval Process
The results of product development, pre-clinical and other non-clinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product. The submission of an NDA is subject to the payment of substantial user fees; a waiver of such fees may be obtained under certain limited circumstances. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant to assure and preserve the product’s identity, strength, quality and purity. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to FDA because the FDA has approximately two months to make a “filing” decision after it the application is submitted. The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review The FDA may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
25
Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP requirements.
After the FDA evaluates an NDA, it will issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application will not be approved in its present form. A Complete Response Letter usually describes the specific deficiencies in the NDA identified by the FDA and may require additional clinical data, such as an additional pivotal Phase 3 trial or other significant and time-consuming requirements related to clinical trials, non-clinical studies or manufacturing. If a Complete Response Letter is issued, the sponsor must resubmit the NDA or, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information are submitted, the FDA may decide that the NDA does not satisfy the criteria for approval.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. In addition, the FDA may require a sponsor to conduct Phase 4 testing, which involves clinical trials designed to further assess a drug’s safety and effectiveness after NDA approval, and may require testing and surveillance programs to monitor the safety of approved products which have been commercialized. The FDA may also place other conditions on approval including the requirement for REMS, to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS. The FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Marketing approval may be withdrawn for non-compliance with regulatory requirements or if problems occur following initial marketing.
The Food and Drug Administration Safety and Innovation Act, or FDASIA, made permanent the Pediatric Research Equity Act, or PREA, which requires a sponsor to conduct pediatric clinical trials for most drugs, for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration. Under PREA, original NDAs and supplements must contain a pediatric assessment unless the sponsor has received a deferral or waiver. The required assessment must evaluate the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The sponsor or FDA may request a deferral of pediatric clinical trials for some or all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the drug is ready for approval for use in adults before pediatric clinical trials are complete or that additional safety or effectiveness data needs to be collected before the pediatric clinical trials begin. The FDA must send a non-compliance letter to any sponsor that fails to submit the required assessment, keep a deferral current or fails to submit a request for approval of a pediatric formulation.
Special FDA Expedited Review and Approval Programs
The FDA has various programs, including Fast Track Designation, accelerated approval, priority review, and breakthrough therapy designation, which are intended to expedite or simplify the process for the development and FDA review of drugs that are intended for the treatment of serious or life threatening diseases or conditions and demonstrate the potential to address unmet medical needs. The purpose of these programs is to provide important new drugs to patients earlier than under standard FDA review procedures.
To be eligible for a Fast Track Designation, the FDA must determine, based on the request of a sponsor, that a product is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address an unmet medical need. The FDA will determine that a product will fill an unmet medical need if it will provide a therapy where none exists or provide a therapy that may be potentially superior to existing therapy based on efficacy or safety factors. The FDA may review sections of the NDA for a fast track product on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, the FDA agrees to accept sections of the NDA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA.
26
The FDA may give a priority review designation to drugs that offer major advances in treatment, or provide a treatment where no adequate therapy exists. A priority review means that the goal for the FDA to review an application is six months, rather than the standard review of ten months under current PDUFA guidelines. Under the new PDUFA agreement, these six and ten month review periods are measured from the “filing” date rather than the receipt date for NDAs for new molecular entities, which typically adds approximately two months to the timeline for review and decision from the date of submission. Most products that are eligible for Fast Track Designation are also likely to be considered appropriate to receive a priority review.
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may be eligible for accelerated approval and may be approved on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require a sponsor of a drug receiving accelerated approval to perform post-marketing studies to verify and describe the predicted effect on irreversible morbidity or mortality or other clinical endpoint, and the drug may be subject to accelerated withdrawal procedures.
Moreover, under the provisions of the FDASIA, a sponsor can request designation of a product candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies are also eligible for accelerated approval. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened. We may explore some of these opportunities for our product candidates as appropriate.
Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual user program fee requirements for any marketed products.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
27
Once an approval of a drug or medical device is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
|
|
• |
restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
|
|
• |
fines, warning letters or holds on post-approval clinical trials; |
|
|
• |
refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals; |
|
|
• |
product seizure or detention, or refusal to permit the import or export of products; or |
|
|
• |
injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs or devices may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
The Hatch-Waxman Amendments
The Drug Price Competition and Patent Term Restoration Act of 1984, known as the Hatch-Waxman Act, added two pathways for FDA drug approval. First, the Hatch-Waxman amendments authorized the FDA to approve an alternative type of NDA under Section 505(b)(2) of the FDCA. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from trials not conducted by or for the applicant and for which the applicant has not obtained a right of reference from the data owner. The applicant may rely upon the FDA’s findings of safety and efficacy for an approved product that acts as the “listed drug.” The FDA may also require 505(b)(2) applicants to perform additional studies or measurements to support the change from the listed drug. The FDA may then approve the new product candidate for all, or some, of the label indications for which the branded reference drug has been approved, as well as for any new indication sought by the 505(b)(2) applicant.
Second, the Hatch-Waxman amendments to the FDCA also established a statutory procedure for submission and FDA review and approval of abbreviated new drug applications, or ANDAs, for generic versions of branded drugs previously approved by the FDA (such previously approved drugs are referred to as “listed drugs”). An ANDA is a comprehensive submission that contains, among other things, data and information pertaining to the active pharmaceutical ingredient, drug product formulation, specifications and stability of the generic drug, as well as analytical methods, manufacturing process validation data and quality control procedures. Premarket applications for generic drugs are termed abbreviated because they generally do not include pre-clinical and clinical data to demonstrate safety and effectiveness. However, a generic manufacturer is typically required to conduct bioequivalence studies of its test product against the listed drug. The bioequivalence studies for orally administered, systemically available drug products assess the rate and extent to which the API is absorbed into the bloodstream from the drug product and becomes available at the site of action. Bioequivalence is established when there is an absence of a significant difference in the rate and extent for absorption of the generic product and the listed drug. For some drugs, other means of demonstrating bioequivalence may be required by the FDA, especially where rate and/or extent of absorption are difficult or impossible to measure. The FDA will approve the generic product as suitable for an ANDA application if it finds that the generic product does not raise new questions of safety and effectiveness as compared to the innovator product. A product is not eligible for ANDA approval if the FDA determines that it is not bioequivalent to the referenced innovator drug, if it is intended for a different use, or if it is not subject to an approved Suitability Petition.
28
In seeking approval for a drug through an NDA, including a 505(b)(2) NDA, applicants are required to list with the FDA certain patents whose claims cover the applicant’s product. Upon approval of an NDA, each of the patents listed in the application for the drug is then published in the Orange Book. Any applicant who files an ANDA seeking approval of a generic equivalent version of a drug listed in the Orange Book or a 505(b)(2) NDA referencing a drug listed in the Orange Book must certify to the FDA that (1) no patent information on the drug product that is the subject of the application has been submitted to the FDA; (2) such patent has expired; (3) the date on which such patent expires; or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of the drug product for which the application is submitted. This last certification is known as a paragraph IV certification. A notice of the paragraph IV certification must be provided to each owner of the patent that is the subject of the certification and to the holder of the approved NDA to which the ANDA or 505(b)(2) application refers. The applicant may also elect to submit a “section viii” statement certifying that its proposed label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent.
If the reference NDA holder and patent owners assert a patent challenge directed to one of the Orange Book listed patents within 45 days of the receipt of the paragraph IV certification notice, the FDA is prohibited from approving the application until the earlier of 30 months from the receipt of the paragraph IV certification expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the applicant. The ANDA or 505(b)(2) application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the branded reference drug has expired.
Marketing Exclusivity
Market exclusivity provisions under the FDCA can delay the submission or the approval of certain marketing applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not approve or even accept for review an abbreviated new drug application, or ANDA, or a NDA submitted under Section 505(b)(2), or 505(b)(2) NDA, submitted by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovative drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. The FDCA alternatively provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for drugs containing the active agent for the original indication or condition of use. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the pre-clinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness. Pediatric exclusivity is another type of marketing exclusivity available in the United States. Pediatric exclusivity provides for an additional six months of marketing exclusivity attached to another period of exclusivity if a sponsor conducts clinical trials in children in response to a written request from the FDA. The issuance of a written request does not require the sponsor to undertake the described clinical trials. In addition, orphan drug exclusivity, as described above, may offer a seven-year period of marketing exclusivity, except in certain circumstances.
U.S. Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any therapeutic product candidate for which we may seek regulatory approval. Sales in the United States will depend in part on the availability of adequate financial coverage and reimbursement from third-party payors, which include government health programs such as Medicare, Medicaid, TRICARE and the Veterans Administration, as well as managed care organizations and private health insurers. Prices at which we or our customers seek reimbursement for our therapeutic product candidates can be subject to challenge, reduction or denial by payors.
29
The process for determining whether a payor will provide coverage for a product is typically separate from the process for setting the reimbursement rate that the payor will pay for the product. A payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be available. Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. In order to obtain coverage and reimbursement for any product that might be approved for marketing, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of any products, which would be in addition to the costs expended to obtain regulatory approvals. Third-party payors may not consider our product candidates to be medically necessary or cost-effective compared to other available therapies, or the rebate percentages required to secure favorable coverage may not yield an adequate margin over cost or may not enable us to maintain price levels sufficient to realize an appropriate return on our investment in drug development.
Healthcare Reform
In the United States and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of drug product candidates, restrict or regulate post-approval activities, and affect the profitable sale of drug product candidates.
Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. In March 2010, the ACA was passed, which substantially changed the way healthcare is financed by both the government and private insurers, and significantly impacts the U.S. pharmaceutical industry. The ACA, among other things: (i) increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extends the rebate program to individuals enrolled in Medicaid managed care organizations; (ii) established an annual, nondeductible fee on any entity that manufactures or imports certain specified branded prescription drugs and biologic agents apportioned among these entities according to their market share in some government healthcare programs; (iii) expanded the availability of lower pricing under the 340B drug pricing program by adding new entities to the program; (iv) increased the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; (v) expanded the eligibility criteria for Medicaid programs; (vi) created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and (vii) established a Center for Medicare Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drugs.
Some of the provisions of the ACA have yet to be implemented, and there have been judicial and Congressional challenges to certain aspects of the ACA. While Congress has not passed comprehensive repeal legislation, bills affecting the implementation of certain taxes under the ACA have been signed into law. The Tax Cuts and Jobs Act of 2017 includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” Additionally, on January 22, 2018, former President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain ACA-mandated fees, including the so-called “Cadillac” tax on certain high-cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax on non-exempt medical devices.
Other legislative changes have been proposed and adopted since the ACA was enacted, including aggregate reductions of Medicare payments to providers of 2% per fiscal year and reduced payments to several types of Medicare providers. Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
30
U.S. Healthcare Fraud and Abuse Laws and Compliance Requirements
Federal and state healthcare laws and regulations restrict business practices in the pharmaceutical industry. The U.S. laws that may affect our ability to operate include:
|
|
• |
the federal Anti-Kickback Statute, which prohibits, among other things, persons from soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
|
|
• |
the federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent; |
|
|
• |
HIPAA, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
|
|
• |
HIPAA, as amended by the federal Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; |
|
|
• |
the federal Physician Payments Sunshine Act, which among other things requires certain manufacturers of drugs, devices, and biologics, that are reimbursable by a federal healthcare program to report annually to the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members; and |
|
|
• |
similar federal laws and state law equivalents of each of the above federal laws. |
Regulation Outside the United States
To the extent that any of our product candidates, once approved, are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws and implementation of corporate compliance programs and reporting of payments or other transfers of value to healthcare professionals.
In order to market our future products in the EEA and many other foreign jurisdictions, we must obtain separate regulatory approvals. More concretely, in the EEA, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of marketing authorizations:
|
|
• |
the Community MA, which is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use of the EMA and which is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, advanced therapy products, and medicinal products containing a new active substance indicated for the treatment certain diseases, such as AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the EU; and |
|
|
• |
National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member State through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. |
31
Under the above described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
Data and Marketing Exclusivity
In the EEA, new products authorized for marketing, or reference products, qualify for eight years of data exclusivity and an additional two years of market exclusivity upon marketing authorization. The data exclusivity period prevents generic or biosimilar applicants from relying on the pre-clinical and clinical trial data contained in the dossier of the reference product when applying for a generic or biosimilar marketing authorization in the EU during a period of eight years from the date on which the reference product was first authorized in the EU. The market exclusivity period prevents a successful generic or biosimilar applicant from commercializing its product in the EU until 10 years have elapsed from the initial authorization of the reference product in the EU. The 10-year market exclusivity period can be extended to a maximum of eleven years if, during the first eight years of those 10 years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. In Japan, medicinal products approved for administration to a patient via a new route of administration qualify for six years of market exclusivity.
Clinical Trials
Clinical trials of medicinal products in the European Union must be conducted in accordance with European Union and national regulations and the International Conference on Harmonization, or ICH, guidelines on GCPs. Additional GCP guidelines from the European Commission, focusing in particular on traceability, apply to clinical trials of advanced therapy medicinal products. If the sponsor of the clinical trial is not established within the European Union, it must appoint an entity within the European Union to act as its legal representative. The sponsor must take out a clinical trial insurance policy, and in most EU countries, the sponsor is liable to provide “no fault” compensation to any study subject injured in the clinical trial.
Prior to commencing a clinical trial, the sponsor must obtain a clinical trial authorization from the competent authority, and a positive opinion from an IEC. The application for a clinical trial authorization must include, among other things, a copy of the trial protocol and an investigational medicinal product dossier containing information about the manufacture and quality of the medicinal product under investigation. Currently, clinical trial authorization applications must be submitted to the competent authority in each EU Member State in which the trial will be conducted. Under the new Regulation on Clinical Trials, which is currently expected to take effect in 2019, there will be a centralized application procedure where one national authority takes the lead in reviewing the application and the other national authorities have only a limited involvement. Any substantial changes to the trial protocol or other information submitted with the clinical trial applications must be notified to or approved by the relevant competent authorities and ethics committees. Medicines used in clinical trials must be manufactured in accordance with cGMP. Other national and European Union-wide regulatory requirements also apply.
Recycling
We started our business in 1976 as a used appliance retailer that reconditioned old appliances to sell in our stores. Under contracts with national and regional retailers of new appliances, we collected the replaced appliance from the retailer’s customer’s residence when one of their stores delivered a new appliance. Any old appliances that we could not sell in our stores were sold to scrap metal processors. In the late 1980s, stricter environmental regulations began to affect the disposal of unwanted appliances and we were no longer able to take appliances that contained hazardous components to a scrap metal processor. At that time, we began to develop systems and equipment to remove the harmful materials so that metal processors would accept the appliance shells for processing. We then offered our services for disposing of appliances in an environmentally sound manner to appliance manufacturers and retailers, waste hauling companies, rental property managers, local governments, and the public. In 1989, we began contracting with electric utility companies to provide turnkey appliance recycling services to support their energy conservation efforts. Since that time, we have provided our services to approximately 400 utilities and other providers of energy efficiency programs throughout North America.
32
We currently have contracts to recycle, or to replace and recycle, major household appliances for approximately 180 utilities across North America. We operate 17 recycling centers in the U.S. and Canada to process and recycle old appliances according to all federal, state, provincial, and local rules and regulations. We use U.S. Environmental Protection Agency (the “EPA”) Responsible Appliance Disposal (“RAD”) Program-compliant methods to remove and properly manage hazardous components and materials, including CFC refrigerants, mercury, polyurethane foam insulation and recyclable materials, such as ferrous and nonferrous metals, plastics, and glass. All of our facilities comply with licensing and permitting requirements, and employees who process appliances receive extensive safety and hazardous materials training.
Major household appliances in the United States include:
|
Refrigerators |
|
Clothes washers |
|
Freezers |
|
Clothes dryers |
|
Ranges/ovens |
|
Room air conditioners |
|
Dishwashers |
|
Dehumidifiers |
|
Microwave ovens |
|
Humidifiers |
Improper disposal of old appliances threatens air, ground, and water resources because many types of major appliances contain substances that can damage the environment. These harmful materials include:
|
|
1. |
Mercury, which easily enters the body through absorption, inhalation, or ingestion, potentially causing neurological damage. Mercury-containing components may be found in older freezers, washers, and ranges. |
|
|
2. |
Chlorofluorocarbon (“CFC”), hydrochlorofluorocarbon, and hydrofluorocarbon refrigerants (collectively, “Refrigerants”), which cause long-term damage to the earth’s ozone layer and may contribute to global climate change. Refrigerators, freezers, room air conditioners, and dehumidifiers commonly contain Refrigerants. |
|
|
3. |
CFCs, having a very high ozone-depletion potential that may also be used as blowing agents in the polyurethane foam insulation of refrigerators and freezers. |
|
|
4. |
Other materials, such as oil, that are harmful when released into the environment. |
The U.S. federal government requires the recovery of refrigerants upon appliance disposal and also regulates the management of hazardous materials found in appliances. Most state and local governments have also enacted laws affecting how their residents dispose of unwanted appliances. For example, many areas restrict landfills and scrap metal processors from accepting appliances unless the units have been processed to remove environmentally harmful materials. As a result, old appliances usually cannot be discarded directly through ordinary solid waste systems.
In addition to these solid waste management and environmental issues, energy conservation is another compelling reason for proper disposal of old appliances. The U.S. Department of Energy’s updated appliance energy efficiency standards that took effect in September 2014 require new refrigerators to be 25-to-30% more efficient than those manufactured only one year earlier. Refrigerators manufactured today use about one-fifth as much electricity as units made in the mid-1970s.
While new refrigerators can save a significant amount of energy in the home, more than 30% of all U.S. households have a second refrigerator in the basement or garage. These units are typically 15-to-25 years old and consume about 750 to 1500 kilowatt-hours per year, driving electric bills up by more than $150 annually per household.
Utilities have become important participants in dealing with energy inefficient appliances as a way of reducing peak demand on their systems and avoiding the capital and environmental costs of adding new generating capacity. To encourage the permanent removal of energy inefficient appliances from use, many electric utility companies sponsor programs through which their residential customers can retire working refrigerators, freezers, and room air conditioners. Utility companies often provide assistance and incentives for consumers to discontinue use of a surplus appliance or to replace their old, inefficient appliances with newer, more efficient models. To help accomplish this, some utilities offer appliance replacement programs for some segments of their customers, through which older model kitchen and laundry appliances are recycled and new highly efficient ENERGY STAR® units are installed.
33
The EPA has been supportive of efforts by electric utilities and other entities that sponsor appliance recycling programs to ensure that the collected units are managed in an environmentally sound manner. In October 2006, the EPA launched RAD, a voluntary partnership program designed to help protect the ozone layer and reduce emissions of greenhouse gases. Through the program, RAD partners use best practices to recover ozone-depleting chemicals and other harmful materials from old refrigerators, freezers, room air conditioners, and dehumidifiers. Because of our appliance recycling expertise, we were active participants in helping to design the RAD program and currently submit annual reports to the EPA to document the environmental benefits our utility customers that are RAD partners have achieved through their recycling programs.
Our wholly-owned subsidiaries in our Recycling segment include, ARCA Canada Inc., a Canadian corporation formed in September 2006, ARCA Recycling, Inc., a California corporation formed in November 1991, and Customer Connexx, LLC, a Nevada limited liability company formed in October 2016 that provides call center services for recycling business.
Technology
On August 18, 2017, in a move to diversify our offering beyond our then-current appliance recycling capabilities, we acquired GeoTraq, which became a wholly-owned subsidiary of the Company. In connection with the acquisition, the Company tendered to the three owners of GeoTraq $200,000, issued to them an aggregate of 288,588 shares of the Company’s Series A Convertible Preferred Stock (which were subsequently exchanged for shares of Series A-1 Convertible Preferred Stock as discussed in this Form 10-K).
GeoTraq is a Mobile Internet of Things (“IoT”) technology company that designs innovative wireless modules that provide Location Based Services (“LBS”) and connect external sensors to the IoT. GeoTraq is planning to manufacture and sell wireless transceiver modules and subscription services that will allow connectivity using publicly available global Mobile IoT networks. GeoTraq addresses the large LBS market segment that is currently under served with existing solutions due to high deployment costs (hardware, service, logistics), limited battery life and large form factors. We believe that there is a large under-served portion of the LBS market that is not addressed by existing solutions. RFID and Wi-Fi require close proximity for asset tracking, while GPS is too bulky and uses too much power for many needs. GeoTraq addresses the white space in-between by designing wireless transceiver modules with technology that provides LBS directly from global Mobile IoT networks. GeoTraq’s technology allows for a substantially lower cost solution, extended service life, a small form factor, and even disposable devices, which we believe can significantly reduce return logistics costs.
GeoTraq applied for and was granted Patent No. 10,182,402, which covers various aspects of operation of its Mobile IoT wireless modules. A description of the patent features and its various claims includes:
|
|
1. |
An apparatus comprising: an interval timer; a power control; a Short Message Service (SMS) packetizer; a geo-locator; a radio frequency (RF) communicator; and a controller and a memory, the memory comprising instructions for the controller to operate the interval timer cooperatively with the power control to cause a transition of the geo-locator from a sleep state to a wake state after a preset defined time interval, and to operate the geo-locator to receive signal strength levels and corresponding cell IDs from a plurality of cellular base stations, and to operate the SMS packetizer to package the signal strength levels and the corresponding cell IDs into a first outgoing SMS message, and to communicate the first outgoing SMS message to a preset address using the RF communicator. |
|
|
2. |
The apparatus of claim 1, further comprising: a subscriber identity module (SIM); and the memory further comprising instructions to block visibility to the SIM by the geo-locator for a limited duration after the transition of the geo-locator from the sleep state to the wake state after the defined time interval. |
|
|
3. |
The apparatus of claim 2, further comprising: the memory further comprising instructions to override a preset floor on the signal strength levels during the limited duration after the transition of the geo-locator from the sleep state to the wake state after the defined time interval. |
|
|
4. |
The apparatus of claim 1, further comprising: the memory further comprising instructions to operate the SMS packetizer to package the signal strength levels with the corresponding cell IDs. |
|
|
5. |
The apparatus of claim 1, further comprising: the memory further comprising instructions to receive a command SMS message via the RF communicator; a parser to extract a time interval command from the received command SMS message; and the memory further comprising instructions to apply the time interval command to the interval timer to set the defined time interval. |
34
|
|
6. |
The apparatus of claim 1, further comprising: the memory further comprising instructions to receive a response SMS message via the RF communicator, the response SMS message being a response to the first outgoing SMS message; a parser to extract geo-locations for cell IDs from the response SMS message; and the memory further comprising instructions to associate the geo-locations for each of the cell IDs from the response message with corresponding cell IDs in the memory. |
|
|
7. |
A method comprising: applying an interval timer to a power control to control power for a subscriber identify module (SIM), a SMS packetizer, a geo-locator, and a radio frequency (RF) communicator after a preset defined time interval; operating the interval timer cooperatively with the power control to cause a transition of the geo-locator from a sleep state to a wake state after the defined time interval; operating the geo-locator to receive signal strength levels and corresponding cell ids from a plurality of cellular base stations; operating the SMS packetizer to package the signal strength levels and the corresponding cell IDs into an outgoing SMS message; and communicating the outgoing SMS message to a preset address using the RF communicator. |
|
|
8. |
The method of claim 7, further comprising: blocking visibility to the SIM by the geo-locator for a limited duration after the transition. |
|
|
9. |
The method of claim 8, further comprising: overriding a preset floor on the signal strength levels during the limited duration after the transition. |
|
|
10. |
The method of claim 7, further comprising: receiving a command SMS message via the RF communicator; extracting a time interval command from the command SMS message; and applying the time interval command to the interval timer to set the defined time interval. |
|
|
11. |
The method of claim 7, further comprising: receiving a response SMS message via the RF communicator in response to the outgoing SMS message; extracting geo-locations for cell IDs from the response SMS message; and associating the geo-locations for each of the cell ids from the response SMS message with corresponding cell IDs in a memory. |
With the GeoTraq acquisition, we expect to have the ability to deploy IoT devices to locate, monitor and track the movement of inventory and other assets and monitor connected sensors. Our GeoTraq subsidiary has not generated any revenue to date, including in the fiscal year ended January 2, 2021.
ApplianceSmart, Inc.
Prior to December 30, 2017, we sold new and out-of-the-box major household appliances in the United States though a chain of Company-owned retail stores operating under the name ApplianceSmart®. On December 30, 2017, we, together with our then-subsidiary, ApplianceSmart, Inc. (“ApplianceSmart”), entered into a Stock Purchase Agreement (the “Stock Purchase Agreement”) with ApplianceSmart Holdings LLC (the “Purchaser”), a wholly-owned subsidiary of Live Ventures Incorporated (Nasdaq: Live), pursuant to which we sold to the Purchaser all of the issued and outstanding shares of capital stock of ApplianceSmart (the “ApplianceSmart Stock”) in exchange for $6.5 million. Effective April 1, 2018, the Purchaser issued the Company a promissory note (the “ApplianceSmart Note”) with a three-year term in the original principal amount of $3.9 million for the balance of the purchase price. ApplianceSmart is guaranteeing the repayment of the ApplianceSmart Note. On December 26, 2018, the ApplianceSmart Note was amended and restated to grant ARCA a security interest in the assets of the Purchaser, ApplianceSmart, and ApplianceSmart Contracting Inc. in exchange for modifying the repayment terms to provide for the payment in full of all accrued interest and principal on April 1, 2021, the maturity date of the ApplianceSmart Note. On March 15, 2019, JanOne entered into subordination agreements with third parties, pursuant to which it agreed to subordinate the payment of indebtedness under the ApplianceSmart Note and its security interest in the assets of ApplianceSmart and other related parties in exchange for receipt of a payment of up to $1.2 million within 15 days of the subordination agreement. On December 9, 2019, ApplianceSmart filed a voluntary petition (the “Chapter 11 Case”) in the United States Bankruptcy Court for the Southern District of New York (the “Bankruptcy Court”), seeking relief under Chapter 11 of Title 11 of the United States Code (the “Bankruptcy Code”). As of January 2, 2021, indebtedness owed by ApplianceSmart to JanOne is approximately $2.9 million. However, JanOne has recorded a full valuation allowance for the entire amount of the indebtedness due to the uncertainty of repayment.
35
Customers and Source of Supply for Recycling and Technology
Recycling: We contract with utility companies or their program administrators and other sponsors of energy efficiency programs to provide a full range of appliance recycling and replacement services to help them achieve their energy savings goals. The contracts usually have terms of one-to-three years, with provisions for renewal at the option of the utility. Under some contracts, we manage all aspects, including advertising of the appliance recycling or replacement program. Under other contracts, we provide only specified services, such as collection and recycling.
GeoTraq: GeoTraq currently has no customers. GeoTraq sources its raw materials, including electronic chips, computers, and software from various third parties. GeoTraq is dependent on a single supplier for its modules.
Principal Products and Services for Recycling and Technology
Our recycling segment generates revenues from two sources: recycling and byproducts. Recycling revenues were generated by charging fees for collecting and recycling appliances for utilities and other sponsors of energy efficiency programs and through the sale of new ENERGY STAR® appliances to utility companies for installation in the homes of a specific segment of their customers. Byproduct revenues were generated by selling scrap materials, such as metal and plastics, from appliances we collected and recycled.
Our technology segment is engaged in the development, design, and ultimately, we expect, the sale of cellular transceiver modules, also known as Mobile IoT modules.
Seasonality for Recycling and Technology
Promotional activities for programs in which the utility sponsor conducts all advertising are generally strong during the second and third calendar quarters, leading to higher customer demand for services during that time period. As a result, we experience a surge in business during the second and third calendar quarters, which generally declines through the fourth and first calendar quarters until advertising activities resume.
Our technology segment did not have any customers at January 2, 2021.
Competition for Recycling and Technology
Recycling:
We generally compete for contracts with several other appliance recycling businesses, energy services management companies, and new-appliance retailers. We also compete with small hauling or recycling companies that are based in the program’s service territory. Many of these companies, including used-appliance dealers that call themselves “appliance recyclers,” resell in the secondary market a percentage of the used appliances they accept for recycling.
We expect our primary competition for appliance recycling and replacement contracts with existing and new customers to come from a variety of sources, including:
|
|
1. |
Existing recycling companies. |
|
|
2. |
Entrepreneurs entering the appliance recycling business. |
|
|
3. |
Major waste hauling companies. |
|
|
4. |
Scrap metal processors. |
|
|
5. |
National and regional new appliance retailers. |
In addition, utility companies and other customers may choose to provide all or some of the services required to operate their appliance recycling and replacement programs internally rather than contracting with outside vendors.
36
Technology
GeoTraq plans on operating in an industry segment that is made of numerous competing technologies designed to connect devices to the IoT. The business’s wireless solution uses IoT based on LTE CAT-M and the newly released NB-IoT protocols that were defined in the GSMA’s (Groupe Speciale Mobile Association) 3GPP Release 13 standard. The Mobile IoT industry utilizes radio spectrum that is licensed to wireless carries by various governmental regulatory agencies around the world. Mobile IoT is extremely competitive and constantly changing as carriers, manufacturers, and solution providers offer innovation to the IoT marketplace. GeoTraq believes there is a large under-served opportunity for “Simple IoT” solutions that significantly reduce the complexity, cycle time and cost of deploying LBS and sensor monitoring solutions. The company’s transceiver modules and associated wireless connectivity subscription service is specifically targeted at accomplishing these objectives.
Government Regulation for Recycling and Technology
Recycling
Federal, state, and local governments regulate appliance collection, recycling, and sales activities. While some requirements apply nationwide, others vary by market. The many laws and regulations that affect appliance recycling include landfill disposal restrictions, hazardous waste management requirements, and air quality standards. For example, the 1990 Amendments to the Clean Air Act prohibit the venting of all Refrigerants while servicing or disposing of appliances.
Each of our recycling facilities maintains the appropriate registrations, permits, and licenses for operating at its location. We register our recycling centers as hazardous waste generators with the EPA and obtain all appropriate regional and local licenses for managing hazardous wastes. Licensed hazardous waste companies transport and recycle or dispose of the hazardous materials we generate. Our collection vehicles and our transportation employees are required to comply with all U.S. Department of Transportation (“DOT”) licensing requirements.
Approximately 30 of ARCA Recycling’s clients participate in the EPA’s voluntary RAD program by committing to employ best environmental practices to reduce emissions of ozone-depleting substances and greenhouse gases through the proper disposal of refrigeration appliances at end of life. We prepare annual RAD reports that quantify the materials collected to submit to EPA on behalf of our clients.
Although we believe that further governmental regulation of the appliance recycling industry could have a positive effect on us, we cannot predict the direction of future legislation. Under some circumstances, for example, further regulation could materially increase our operational costs or reduce environmental requirements for disposing of appliances at end of life. In addition, under some circumstances we may be subject to contingent liabilities because we handle hazardous materials. We believe we are in compliance with all government regulations regarding the handling of hazardous materials, and we have environmental insurance to mitigate the impact of any potential contingent liability.
Technology
GeoTraq’s Mobile IoT modules utilize low-power wireless transmitters that emit RF energy waves, which are subject to regulation by the Federal Communications Commission (“FCC”) and may be subject to regulation by other domestic and international agencies. GeoTraq believes that FCC rules Part 15, Part 20, Part 22, Part 24, and Part 27 may apply to the company’s products. GeoTraq believes that its products are safe and will utilize FCC accredited testing laboratories to verify and certify that its modules comply with all required regulatory requirements. In addition, GeoTraq intends to seek and obtain necessary licenses and permits from the FCC and other regulatory agencies as required by law.
Employees
On January 2, 2021, we had 179 employees, of which 172 were full-time employees.
37
You should carefully consider the risks described below with respect to an investment in our shares. If any of the following risks actually occur, our business, financial condition, operating results or cash provided by operations could be materially harmed. As a result, the trading price of our common stock could decline, and you might lose all or part of your investment. When evaluating an investment in our common stock, you should also refer to the other information in this Form 10-K, including our consolidated financial statements and related notes.
Risks Relating to Our Business Generally
Our results of operations may be negatively impacted by the coronavirus outbreak.
In December 2019, the 2019 novel coronavirus (COVID-19) surfaced in Wuhan, China. The World Health Organization declared a global emergency on January 30, 2020, and most countries initiated travel restrictions limiting travel to other countries and lock-downs within their borders. While various vaccines have recently been introduced into the marketplace, the impacts of variant strains of the COVID-19 virus is still unknown.
The widespread health crisis has adversely affected the global economy, resulting in an economic downturn that could impact demand for our products.
To date, the outbreak had a material adverse impact on our operations. For example, several customers in our appliance recycling and appliance replacement business have previously suspended our ability to pick up and or replace their customers’ appliances resulting in decreased revenues for both recycling and replacement business. The future impact of the outbreak is highly uncertain and cannot be predicted and there is no assurance that the outbreak will not have another material adverse impact on the future results of the Company. The extent of the impact, if any, will depend on future developments, including actions taken to contain the coronavirus.
A key task for the Company in 2021 is to begin late-stage clinical development with its pharmaceutical product, JAN101. However, the COVID-19 pandemic significantly impacted clinical trials in 2020, delaying recruitment in most non-COVID-19 clinical trials and even eliminating recruitment in some trials. While clinical sites have largely resumed conducting non-COVID-19 clinical trials, the backlog of subjects may adversely affect our ability to recruit for its trial, leading to longer and more expensive trials. In addition, the unknown effectiveness of the COVID-19 vaccines, particularly concerning variant strains of COVID-19, could lead to clinical sites terminating patient recruitment again during the course of the study.
If we fail to implement our biopharmaceutical business strategy or if our biopharmaceutical business strategy is ineffective, our financial performance could be materially and adversely affected.
Our future financial performance and success are dependent in large part upon the effectiveness of our new biopharmaceutical business strategy and our ability to implement our biopharmaceutical business strategy successfully. Implementation of our strategy will require effective management of our operational, financial, and human resources and will place significant demands on those resources. There are risks involved in pursuing our strategy, including those under the caption “- Risks Relating to Our Biotechnology Segment”. In addition to the risks set forth above, effectiveness of and the successful implementation of our business strategy could also be affected by a number of factors beyond our control, such as increased competition, legal developments, government regulation, general economic conditions, increased operating costs or expenses and changes in industry trends. We may decide to alter or discontinue certain aspects of our business strategy at any time. If we are not able to implement our business strategy successfully, our long-term growth and profitability may be adversely affected. Even if we are able to implement some or all of the initiatives of our business strategy successfully, our operating results may not improve and could decline substantially.
38
We may be unable to complete the disposition of our recycling business.
On February 19, 2021, we, together with our subsidiaries (a) ARCA Recycling, Inc., a California corporation (“ARCA”), and (b) Customer Connexx LLC, a Nevada limited liability company (“Connexx”), entered into an Asset Purchase Agreement (the “Purchase Agreement”) with (i) ARCA Affiliated Holdings Corporation, a Delaware corporation, (ii) ARCA Services Inc., a Delaware corporation, and (iii) Connexx Services Inc, a Delaware corporation (collectively, the “Buyers”), pursuant to which the Buyers agreed to acquire substantially all of the assets, and assume certain liabilities, of ARCA and Connexx (the “Disposition Transaction”). The principal of the Buyers is Virland A. Johnson, our Chief Financial Officer. The Disposition Transaction is expected to be consummated on or before August 18, 2021 and is subject to certain customary closing conditions, which makes its completion and timing uncertain. Accordingly, there can be no assurance that the Disposition Transaction will be consummated on the anticipated schedule or at all. If we are unable to complete the Disposition Transaction, we may be required to identify a new purchaser and renegotiate the sale of the recycling business, and any such new sale would also be subject to new regulatory and other conditions. Such renegotiation and conditions and the process of obtaining regulatory approvals could have the effect of delaying or impeding consummation of the sale of the recycling business. A delay or failure to sell the recycling business could have a material adverse effect on our business, financial position or results of operations.
We have identified and disclosed in this Form 10-K material weaknesses in our internal control over financial reporting. If we are not able to remediate these material weaknesses and maintain an effective system of internal controls, we may not be able to accurately or timely report our financial results, which could cause our stock price to fall or result in our stock being delisted.
We need to devote significant resources and time to comply with the requirements of the Sarbanes-Oxley Act of 2002 (“Sarbanes-Oxley”) with respect to internal control over financial reporting. In addition, Section 404 under Sarbanes-Oxley requires that we assess the design and operating effectiveness of our controls over financial reporting, which are necessary for us to provide reliable and accurate financial reports.
As reported in Part II – Item 9A, Controls and Procedures, there were material weaknesses in our internal controls over financial reporting at January 2, 2021. Specifically, management noted the following material weaknesses in internal control when conducting their evaluation of internal control as of January 2, 2021: (1) Insufficient information technology general controls (“ITGC”) and segregation of duties. It was noted that people who were negotiating a contract, were also involved in approving invoices without proper oversight. Additional controls and procedures are necessary and are being implemented to have check and balance on significant transactions and governance with those charged with governance authority. (2) Inadequate control design or lack of sufficient controls over significant accounting processes. The cutoff and reconciliation procedures were not effective with certain accrued and deferred expenses. (3) Insufficient assessment of the impact of potentially significant transactions, and (4) Insufficient processes and procedures related to proper recordkeeping of agreements and contracts In addition, contract to invoice reconciliation was not effective with certain transportation service providers. As part of its remediation plan, processes and procedures have been implemented to help ensure accruals and invoices are reviewed for accuracy and properly recorded in the appropriate period.
We expect our systems and controls to become increasingly complex to the extent that we integrate acquisitions and as our business grows. To effectively manage our company today and this anticipated complexity, we need to remediate these material weaknesses and continue to improve our operational, financial, and management controls and our reporting systems and procedures. Any failure to remediate these material weaknesses and implement required new or improved controls, or difficulties encountered in the implementation or operation of these controls, could harm our operating results or cause us to fail to meet our financial reporting obligations, which could adversely affect our business and jeopardize our listing on the Nasdaq Capital Market, either of which would harm our stock price.
39
Risks Relating to Our Biotechnology Segment
Our biotechnology business has a limited operating history.
Our biotechnology business was started in September 2019 and has limited operating history. We have not commenced revenue-producing operations. To date, our biotechnology-related operations have consisted of preliminary research and development, and characterization and testing of SR TV1001 (now known as JAN101), our initial product candidate. Our limited operating history makes it difficult for potential investors to evaluate our technology or the prospective operations of our biotechnology business. You should consider the prospects of our biotechnology business in light of the costs, uncertainties, delays and difficulties frequently encountered by companies in the early stages of development, especially clinical-stage biopharmaceutical businesses such as ours. Potential investors should carefully consider the risks and uncertainties that a biotechnology business with a limited operating history faces. In particular, potential investors should consider that we may be unable to (i) successfully implement or execute the business plan of our biotechnology business, or that our biotechnology business plan is sound; (ii) successfully complete clinical trials and obtain regulatory approval for the marketing of our product candidate; (iii) successfully demonstrate a favorable differentiation between our product candidate and the current products on the market; (iv) successfully manufacture of our clinical drug product and establish a commercial drug supply; (v) secure market exclusivity and/or adequate intellectual property protection for our product candidate; and (vi) raise sufficient funds in the capital markets to effectuate our biotechnology business plan, including product and clinical development, regulatory approval, and commercialization for our product candidate.
Our business model is entirely dependent on certain patent rights licensed to us from the Licensors (as defined below), and the loss of those license rights would, in all likelihood, cause our business, as presently contemplated, to fail.
In November 2019, UAB Research Foundation (“UABRF”), TheraVasc, Inc. (“TheraVasc”), and the Board of Supervisors of Louisiana State University and Agricultural and Mechanical College, acting on behalf of LSU Health Sciences Center at Shreveport (“LSU Health Shreveport”, together with UABRF and TheraVasc, the “Licensors”), granted us an exclusive worldwide, royalty bearing license to the patent rights for SR TV1001 (now known as JAN101) in the negotiated fields of use. The patent license agreement requires us to pay royalties and milestone payments and conform to a variety of covenants and agreements, and in the event of our breach of the agreement, the Licensors may elect to terminate the agreement. As of the date of this Form 10-K, we believe we are in compliance with the patent license agreement and consider our relationship with the Licensors to be excellent.
We will be completely dependent on third parties to manufacture our product candidate, and the commercialization of our product candidate could be halted, delayed, or made less profitable if those third parties fail to obtain manufacturing approval from the FDA or comparable foreign regulatory authorities, fail to provide us with sufficient quantities of our product candidates or fail to do so at acceptable quality levels or prices.
We do not currently have, nor do we plan to acquire, the capability or infrastructure to manufacture our drug candidate for use in our clinical trials or for commercial sales, if any. As a result, we will be obligated to rely on contract manufacturers, when we conduct clinical trials and if and when any of our product candidates are approved for commercialization. In January 2020, we entered into a Master Agreement for Development, Manufacturing and Supply with CoreRx Inc. (“CoreRx”), pursuant to which CoreRx has agreed to provide to us certain product testing, development, and clinical manufacturing services. We have not entered into agreements with any contract manufacturers for commercial supply and may not be able to engage contract manufacturers for commercial supply of any of our product candidates on favorable terms to us, or at all, should the need arise.
In a previous clinical trial, the manufacture of JAN101 by a different manufacturing company resulted in product that demonstrated initial instability that led to the product being out-of-specification. While the FDA allowed the trial to continue, there is no guarantee that if the product manufactured by CoreRx is similarly unstable, the FDA will allow us to continue to develop the product. Even if the product manufactured by CoreRx is stable, the FDA may require additional studies to confirm the stability of the product, increasing development cost and times.
40
The facilities used by CoreRx to manufacture our product candidate must be approved by the FDA or comparable foreign regulatory authorities. Such approvals are subject to inspections that will be conducted after we submit an NDA to the FDA or their equivalents to other relevant regulatory authorities. We will not control the manufacturing process of our product candidates and will be completely dependent on our contract manufacturing partners for compliance with cGMPs, for manufacture of both active drug substances and finished drug products. These cGMP regulations cover all aspects of the manufacturing, testing, quality control, storage, distribution and record keeping relating to our product candidates. If our contract manufacturers do not successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others, we will not be able to secure or maintain regulatory approval for product made at their manufacturing facilities. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, manufacture, obtain regulatory approval for or market our product candidates, if approved. Likewise, we could be negatively impacted if any of our contract manufacturers elect to discontinue their business relationship with us.
Our contract manufacturer will be subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies for compliance with cGMPs and similar regulatory requirements. We will not have control over our contract manufacturer’s compliance with these regulations and standards. Failure by our contract manufacturer to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure to grant approval to market our product candidate, delays, suspensions or withdrawals of approvals, inability to supply product, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect our biotechnology business. In addition, we will not have control over the ability of our contract manufacturer to maintain adequate quality control, quality assurance and qualified personnel. Failure by our contract manufacturer to comply with or maintain any of these standards could adversely affect our ability to develop, manufacture, obtain regulatory approval for or market our product candidate, if approved.
Our manufacturer must obtain the active pharmaceutical ingredient, or API, from a third party. A number of groups manufacturer our API, however, some of these are manufactured as a food product, and others, while manufactured under GMP do not have the required Drug Master File on file with the FDA. CoreRx identified API from Merck KGaA for use in the current production of clinical grade JAN101. At the time of the manufacture of the API, the product met the specifications outlined in both the drug substance monographs for Europe and the US. However, subsequent to the manufacture of the API, the US monograph was changed in the US Pharmacopeia (USP) and while most of the test conform, Merck KGaA was unable to complete two of the new testing requirements. Although the two tests are not considered safety issues and do not impact the quality of the product, there is no guarantee the FDA will approve the product for clinical trials if the two tests are not completed, which could delay the ability to start the Phase 2b clinical trial as planned. Identifying an analytical laboratory to perform the two tasks may be difficult and could require development and validation of the tests, adding both time and costs to the Company. In addition, there is no guarantee that once developed, the product will meet the specifications as outlined in the USP. Even if the FDA allows the current product to be used in the Phase 2b clinical trial, there is no guarantee that the FDA will allow further clinical work with the product or commercialization of the product until it is shown to conform to USP standards. We may be required to work with the API manufacturer to file the appropriate documents and there is no guarantee that the FDA will approve the filing. This could necessitate additional funding to hire an API manufacturer and produce the product under GMP with all necessary filings.
If, for any reason, these third parties are unable or unwilling to perform, we may not be able to locate alternative manufacturers or formulators or enter into favorable agreements with them and we cannot be certain that any such third parties will have the manufacturing capacity to meet future requirements. If these manufacturers or any alternate manufacturer of finished drug product experiences any significant difficulties in its respective manufacturing processes for APIs, or finished products or should cease doing business with us for any reason, we could experience significant interruptions in the supply of any of our product candidates or may not be able to create a supply of our product candidates at all. Were we to encounter manufacturing difficulties, our ability to produce a sufficient supply of any of our product candidates might be negatively affected. Our inability to coordinate the efforts of our third-party manufacturing partners, or the lack of capacity available at our third-party manufacturing partners, could impair our ability to supply any of our product candidates at required levels. Because of the significant regulatory requirements that we would need to satisfy in order to qualify a new bulk drug substance or finished product manufacturer, if we face these or other difficulties with our then current manufacturing partners, we could experience significant interruptions in the supply of any of our product candidates if we decided to transfer the manufacture of any of our product candidates to one or more alternative manufacturers in an effort to deal with such difficulties.
41
CoreRx currently serves as our sole manufacturer of JAN101. As CoreRx also manufactures other products, there can be no guarantee that CoreRx will have the capacity to manufacture additional clinical product for us in a timely manner when required, which could lead to significant delays in initiating other clinical studies. CoreRx will unlikely have the capacity to manufacture the amount of product needed, when and if JAN101 is approved for marketing. This could necessitate identifying additional manufacturer(s) who may have problems replicating the manufacturing process developed at CoreRx. In addition, the increase in quantities required for commercialization of the product, if commercialize occurs, could require modifying the manufacturing process to produce larger quantities of tablets more efficiently. Such modifications of the manufacturing process, if even possible, could results in significantly delays in the delivery of the product.
Any manufacturing problem or the loss of our contract manufacturer could be disruptive to our operations and result in development delays and lost sales. Additionally, we will rely on third parties to supply the raw materials needed to manufacture our product candidates. Any such reliance on suppliers may involve several risks, including a potential inability to obtain critical materials and reduced control over production costs, delivery schedules, reliability and quality. Any unanticipated disruption to the operation of one of our contract manufacturers caused by problems with suppliers could delay shipment of any of our product candidates, increase our cost of goods sold and result in lost sales.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We will face a potential risk of product liability as a result of the clinical testing of our product candidate. For example, we may be sued if any product we develop, including our product candidate, or any materials that we use in our product candidate, allegedly causes injury or is found to be otherwise unsuitable during product testing and manufacturing. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability, and a breach of warranties. In the U.S., claims could also be asserted against us under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidate. Even successful defense of these claims would require us to employ significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in, among other things (i) decreased demand for our product candidate or any future products that we may develop; (ii) failure to obtain regulatory approval for our product candidate; (iii) withdrawal of participants in our clinical trials; (iv) substantial monetary awards to trial participants or patients; (v) product recalls, withdrawals or labeling, marketing or promotional restrictions; and (vi) the inability to commercialize our product candidate. As of the date of this Form 10-K, we do not carry product liability insurance.
The success of our biotechnology business is entirely dependent on our ability to obtain the marketing approval for our product candidate by the FDA and the regulatory authorities in foreign jurisdictions in which we intend to market our product candidate, of which there can be no assurance.
We are not permitted to market our product candidate as prescription pharmaceutical products in the United States until we receive approval of an NDA from the FDA, or in any foreign countries until we receive the requisite approval from such countries. In the United States, the FDA generally requires the completion of clinical trials of each drug to establish its safety and efficacy and extensive pharmaceutical development to ensure its quality before an NDA is approved. Of the large number of drugs in development, only a small percentage result in the submission of an NDA to the FDA and even fewer are eventually approved for commercialization. As of the date of this Form 10-K, we have not submitted an NDA to the FDA or comparable applications to other regulatory authorities for any of our product candidates.
Because of the clinical trial history of JAN101, we believe that our initial drug product candidate will qualify for FDA approval through the FDA’s 505(b)(2) regulatory pathway and in corresponding regulatory paths in other foreign jurisdictions. Notwithstanding the use of the FDA’s 505(b)(2) regulatory pathway, we will be required to conduct Phase IIb and Phase III studies prior to filing for marketing approval of our product candidate.
42
Our success depends on our receipt of the regulatory approvals described above, and the issuance of such regulatory approvals is uncertain and subject to a number of risks, including the following: (i) the results of toxicology studies may not support the filing of an NDA for our product candidates; (ii) the FDA may require additional pharmacokinetic studies with JAN101, including studies with food, prior to allowing the Company to conduct Phase IIb and Phase III trials; (iii) the FDA or comparable foreign regulatory authorities or Institutional Review Boards, or IRB, may disagree with the design or implementation of our clinical trials; (iv) we may not be able to provide acceptable evidence of our product candidate’s safety and efficacy; (v) the results of our clinical trials may not be satisfactory or may not meet the level of statistical or clinical significance required by the FDA, European Medicines Agency, or EMA, or other regulatory agencies for us to receive marketing approval for our product candidate; (vi) the dosing of our product candidate in a particular clinical trial may not be at an optimal level; (vii) patients in our clinical trials may suffer adverse effects for reasons that may or may not be related to our product candidate; (viii) the data collected from clinical trials may not be sufficient to support the submission of an NDA or other submission or to obtain regulatory approval in the United States or elsewhere; (ix) the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and (x) the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval of our product candidate.
The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon, among other things, the type, complexity, and novelty of the product candidates involved, the jurisdiction in which regulatory approval is sought and the substantial discretion of the regulatory authorities. Changes in regulatory approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for a submitted product application may cause delays in the approval or rejection of an application. Regulatory approval obtained in one jurisdiction does not necessarily mean that a product candidate will receive regulatory approval in all jurisdictions in which we may seek approval, but the failure to obtain approval in one jurisdiction may negatively impact our ability to seek approval in a different jurisdiction. Failure to obtain regulatory approval for our product candidate for the foregoing, or any other reasons, will prevent us from commercializing our product candidate, and our ability to generate revenue will be materially impaired.
Clinical testing is expensive, is difficult to design and implement, can take many years to complete and is uncertain as to outcome.
Our business model depends in part on the successful development, regulatory approval and commercialization of our product candidate, which may never occur. Our product candidate is in the early stages of development and as of the date of this Form 10-K we have not progressed our product candidate beyond early clinical studies designed only to show safety. Three INDs have previously been submitted by previous licensees/assignees of JAN101 and accepted by the FDA. These INDs were transferred to JanOne in 2020. Even though the INDs were transferred to us, the FDA may still require additional work prior to re-initiation of clinical trials. If we do not obtain such approvals to re-initiate trials as presently planned, the time in which we expect to commence clinical programs for any product candidate will be extended and such extension will increase our expenses, delay our potential receipt of any revenues, and increase our need for additional capital. Moreover, there is no guarantee that we will receive approval to commence human clinical trials or, if we do receive approval, that our clinical trials will be successful or that we will continue clinical development in support of an approval from the FDA or comparable foreign regulatory authorities for any indication. We note that most product candidates never reach the clinical development stage and even those that do commence clinical development have only a small chance of successfully completing clinical development and gaining regulatory approval. Success in early phases of pre-clinical and clinical trials does not ensure that later clinical trials will be successful, and interim results of a clinical trial do not necessarily predict final results. A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates. Therefore, our business currently depends entirely on the successful development, regulatory approval and commercialization of our product candidates, which may never occur.
43
Even if we receive regulatory approval for our product candidate, we may not be able to successfully commercialize the product and the revenue that we generate from its sales, if any, may be limited.
If approved for marketing, the commercial success of our product candidate will depend upon the product’s acceptance by the medical community, including physicians, patients, and health care payors. The degree of market acceptance for our product candidate will depend on a number of factors, including (i) demonstration of clinical safety and efficacy; (ii) relative convenience, dosing burden and ease of administration; (iii) the prevalence and severity of any adverse effects; (iv) the willingness of physicians to prescribe our product candidate, and the target patient population to try new therapies; (v) efficacy of our product candidate compared to competing products; (vi) the introduction of any new products that may in the future become available targeting indications for which our product candidate may be approved; (vii) new procedures or therapies that may reduce the incidences of any of the indications in which our product candidate may show utility; (viii) pricing and cost-effectiveness; (ix) the inclusion or omission of our product candidate in applicable guidelines; (x) the effectiveness of our own or any future collaborators’ sales and marketing strategies; (xi) limitations or warnings contained in approved labeling from regulatory authorities; (xii) our ability to obtain and maintain sufficient third-party coverage or reimbursement from government health care programs, including Medicare and Medicaid, private health insurers and other third-party payors or to receive the necessary pricing approvals from government bodies regulating the pricing and usage of therapeutics; and (xiii) the willingness of patients to pay out-of-pocket in the absence of third-party coverage or reimbursement or government pricing approvals.
If our product candidate is approved but does not achieve an adequate level of acceptance by physicians, health care payors, and patients, our biotechnology business may not generate sufficient revenue to cover costs. Our efforts to educate the medical community and third-party payors on the benefits of our product candidate may require significant resources and may never be successful.
In addition, even if we obtain regulatory approvals, the timing or scope of any approvals may prohibit or reduce our ability to commercialize our product candidate successfully. For example, if the approval process takes too long, we may miss market opportunities and give other companies the ability to develop competing products or establish market dominance. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that renders our product candidate not commercially viable. For example, regulatory authorities may approve our product candidate for fewer or more limited indications than we request, may not approve the price we intend to charge for our product candidate, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve any of our product candidates with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that indication. Further, the FDA or comparable foreign regulatory authorities may place conditions on approvals or require risk management plans or a Risk Evaluation and Mitigation Strategy, or REMS, to assure the safe use of the drug. Moreover, product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following the initial marketing of the product. Any of the foregoing scenarios could materially harm the commercial success of our product candidate.
Even if we obtain marketing approval for our product candidate, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense. Additionally, our product candidate could be subject to labeling and other restrictions and withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our product candidate.
Even if we obtain regulatory approval for our product candidate for an indication, the FDA or foreign equivalent may still impose significant restrictions on its indicated uses or marketing or the conditions of approval, or impose ongoing requirements for potentially costly and time-consuming post-approval studies, including Phase 4 clinical trials, and post-market surveillance to monitor safety and efficacy. Our product candidate will also be subject to ongoing regulatory requirements governing the manufacturing, labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, recordkeeping and reporting of adverse events and other post-market information. These requirements include registration with the FDA, as well as continued compliance with current good clinical practices regulations for any clinical trials that we conduct post-approval. In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current cGMPs, requirements relating to quality control, quality assurance and corresponding maintenance of records and documents.
The FDA has the authority to require a REMS as part of an NDA or after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria or requiring patient testing, monitoring and/or enrollment in a registry.
44
With respect to sales and marketing activities related to our product candidate, advertising and promotional materials must comply with FDA rules in addition to other applicable federal, state and local laws in the United States and similar legal requirements in other countries. In the United States, the distribution of product samples to physicians must comply with the requirements of the U.S. Prescription Drug Marketing Act. Application holders must obtain FDA approval for product and manufacturing changes, depending on the nature of the change. We may also be subject, directly or indirectly through our customers and partners, to various fraud and abuse laws, including, without limitation, the U.S. Anti-Kickback Statute, U.S. False Claims Act, and similar state laws, which impact, among other things, our proposed sales, marketing, and scientific/educational grant programs. If we participate in the U.S. Medicaid Drug Rebate Program, the Federal Supply Schedule of the U.S. Department of Veterans Affairs, or other government drug programs, we will be subject to complex laws and regulations regarding reporting and payment obligations. All of these activities are also potentially subject to U.S. federal and state consumer protection and unfair competition laws. Similar requirements exist in many of these areas in other countries.
In addition, if our product candidate is approved for a particular indication, our product labeling, advertising and promotion would be subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. If we receive marketing approval for our product candidate, physicians may nevertheless legally prescribe our product to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability and government fines. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees of permanent injunctions under which specified promotional conduct is changed or curtailed.
If we or a regulatory agency discover previously unknown problems with a product candidate, such as adverse events of unanticipated severity or frequency, problems with the facility where the product is manufactured, or we or our manufacturers fail to comply with applicable regulatory requirements, we may be subject to the following administrative or judicial sanctions: (i) restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; (ii) issuance of warning letters or untitled letters; (iii) clinical holds; (iv) injunctions or the imposition of civil or criminal penalties or monetary fines; (v) suspension or withdrawal of regulatory approval; (vi) suspension of any ongoing clinical trials; (vii) refusal to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product license approvals; (viii) suspension or imposition of restrictions on operations, including costly new manufacturing requirements; or (ix) product seizure or detention or refusal to permit the import or export of product. The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidate and generate revenue. Adverse regulatory action, whether pre- or post-approval, can also potentially lead to product liability claims and increase our product liability exposure.
Obtaining and maintaining regulatory approval of our product candidate in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidate in other jurisdictions.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, but a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants marketing approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from those in the United States, including additional preclinical studies or clinical trials, as clinical studies conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval.
45
Current and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for our product candidate, restrict, or regulate post-approval activities and affect our ability to profitably sell our product candidate. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We do not know whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidate, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
Any termination or suspension of, or delays in the commencement or completion of, any necessary studies of any of our product candidate for any indications could result in increased costs to us, delay or limit our ability to generate revenue and adversely affect our commercial prospects.
The commencement and completion of clinical studies can be delayed for a number of reasons, including delays related to: (i) the FDA or a comparable foreign regulatory authority failing to grant permission to proceed and placing the clinical study on hold; (ii) subjects for clinical testing failing to enroll or remain enrolled in our trials at the rate we expect; (iii) a facility manufacturing any of our product candidates being ordered by the FDA or other government or regulatory authorities to temporarily or permanently shut down due to violations of cGMP requirements or other applicable requirements, or cross-contaminations of product candidates in the manufacturing process; (iv) any changes to our manufacturing process that may be necessary or desired; (v) subjects choosing an alternative treatment for the indications for which we are developing our product candidates, or participating in competing clinical studies; (vi) subjects experiencing severe or unexpected drug-related adverse effects; (vii) reports from clinical testing on similar technologies and products raising safety and/or efficacy concerns; (viii) third-party clinical investigators losing their license or permits necessary to perform our clinical trials, not performing our clinical trials on our anticipated schedule or employing methods consistent with the clinical trial protocol, cGMP requirements, or other third parties not performing data collection and analysis in a timely or accurate manner; (ix) inspections of clinical study sites by the FDA, comparable foreign regulatory authorities, or IRBs finding regulatory violations that require us to undertake corrective action, result in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study, or that prohibit us from using some or all of the data in support of our marketing applications; (x) third-party contractors becoming debarred or suspended or otherwise penalized by the FDA or other government or regulatory authorities for violations of regulatory requirements, in which case we may need to find a substitute contractor, and we may not be able to use some or any of the data produced by such contractors in support of our marketing applications; (xi) one or more IRBs refusing to approve, suspending or terminating the study at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; (xii) reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; (xiii) deviations of the clinical sites from trial protocols or dropping out of a trial; (xiv) adding new clinical trial sites; (xv) the inability of the CRO to execute any clinical trials for any reason; and (xvi) government or regulatory delays or “clinical holds” requiring suspension or termination of a trial.
Product development costs for any of our product candidates will increase if we have delays in testing or approval or if we need to perform more or larger clinical studies than planned. Additionally, changes in regulatory requirements and policies may occur and we may need to amend study protocols to reflect these changes. Amendments may require us to resubmit our study protocols to the FDA, comparable foreign regulatory authorities, and IRBs for reexamination, which may impact the costs, timing or successful completion of that study. If we experience delays in completion of, or if we, the FDA or other regulatory authorities, the IRB, or other reviewing entities, or any of our clinical study sites suspend or terminate any of our clinical studies of any of our product candidates, its commercial prospects may be materially harmed and our ability to generate product revenues will be delayed. Any delays in completing our clinical trials will increase our costs, slow down our development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, termination or suspension of, or a delay in the commencement or completion of, clinical studies may also ultimately lead to the denial of regulatory approval of our product candidates. In addition, if one or more clinical studies are delayed, our competitors may be able to bring competing products to market before we do, and the commercial viability of any of our affected product candidates could be significantly reduced.
46
Third-party coverage and reimbursement and health care cost containment initiatives and treatment guidelines may constrain our future revenues.
Our ability to successfully market our product candidate will depend in part on the level of reimbursement that government health administration authorities, private health coverage insurers and other organizations provide for the cost of our product candidate and related treatments. Countries in which our product candidate is sold through reimbursement schemes under national health insurance programs frequently require that manufacturers and sellers of pharmaceutical products obtain governmental approval of initial prices and any subsequent price increases. In certain countries, including the United States, government-funded and private medical care plans can exert significant indirect pressure on prices. We may not be able to sell our product candidate profitably if adequate prices are not approved or coverage and reimbursement is unavailable or limited in scope. Increasingly, third-party payors attempt to contain health care costs in ways that are likely to impact the development of our product including: (i) failing to approve or challenging the prices charged for health care products; (ii) introducing reimportation schemes from lower priced jurisdictions; (iii) limiting both coverage and the amount of reimbursement for new therapeutic products; (iv) denying or limiting coverage for products that are approved by the regulatory agencies but are considered to be experimental or investigational by third-party payors; and (v) refusing to provide coverage when an approved product is used in a way that has not received regulatory marketing approval.
It is difficult and costly to protect our intellectual property rights, and we cannot ensure the protection of these rights.
Our success depends on successfully blocking others from developing and commercializing similar products. As a repurposed drug, our API has previously been approved for other indications, none of which currently represent a threat to our product, and therefore cannot be protected. We will rely on our method of use and oral formulation patents to protect our product, which may also put our product at risk from companies developing oral formulations using the same API for other indications. Even though our patents provide protection for specific uses, we will not be able to prevent other companies from developing the same API for other uses. If a similar dose, formulation and route of administration is developed for another indication by a different company, we cannot guarantee that the product they market for the other indication will not be prescribed off-label by doctors or filled by pharmacists for use in indications our patents cover and that if less expensive, would not negatively affect our sales, if our product is ultimately approved by the FDA
The degree of future protection afforded by the patent rights licensed to us is uncertain, because legal means afford only limited protection and may not adequately protect our rights, permit us to gain or keep our competitive advantage, or provide us with any competitive advantage at all. We cannot be certain that any patent application owned by a third party will not have priority over patent applications in which we hold license rights or that we will not be involved in interference, opposition or invalidity proceedings before United States or foreign patent offices.
Additionally, if the Licensors were to initiate legal proceedings against a third party to enforce a patent covering our product candidate, the defendant could counterclaim that such patent is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include alleged failures to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for unenforceability assertions include allegations that someone connected with prosecution of the patent withheld relevant information from the United States Patent and Trademark Office, or the U.S. PTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review and equivalent proceedings in foreign jurisdictions, e.g., opposition proceedings. Such proceedings could result in revocation or amendment of the Licensors’ patents in such a way that they no longer cover our product candidate or competitive products. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot be certain that there is no invalidating prior art, of which the Licensors and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on any of our product candidates. Such a loss of patent protection would have a material adverse impact on our business.
47
In the future, we may rely on know-how and trade secrets to protect technology, especially in cases in which we believe patent protection is not appropriate or obtainable. However, know-how and trade secrets are difficult to protect. While we intend to require employees, academic collaborators, consultants and other contractors to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary or licensed information. Typically, research collaborators and scientific advisors have rights to publish data and information in which we may have rights. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts are sometimes less willing to protect trade secrets than patents. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
If we fail to obtain or maintain patent protection or trade secret protection for our product candidates or our technologies, third parties could use our proprietary information, which could impair our ability to compete in the market and adversely affect our ability to generate revenues and attain profitability.
It is difficult and costly to block others from developing similar products for other indications, and we cannot ensure that these products will not be less expensive and thus be prescribed off-label by physicians for use in our indications.
Our success depends on successfully blocking others from developing and commercializing similar products. As a repurposed drug, our API has previously been approved for other indications, none of which currently represent a threat to our product, and therefore cannot be protected. We will rely on our method of use and oral formulation patents to protect our product, which may also put our product at risk from companies developing oral formulations using the same API for other indications. Even though our patents provide protection for specific uses, we will not be able to prevent other companies from developing the same API for other uses. If a similar dose, formulation and route of administration is developed for another indication by a different company, we cannot guarantee that the product they market for the other indication will not be prescribed off-label by doctors or filled by pharmacists for use in indications our patents cover and that if less expensive, would not negatively affect our sales, if our product is ultimately approved by the FDA.
Our product candidate may infringe the intellectual property rights of others, which could increase our costs and delay or prevent our development and commercialization efforts.
Our success depends in part on avoiding infringement of the proprietary technologies of others. The pharmaceutical industry has been characterized by frequent litigation regarding patent and other intellectual property rights. Identification of third-party patent rights that may be relevant to our proprietary technology is difficult because patent searching is imperfect due to differences in terminology among patents, incomplete databases and the difficulty in assessing the meaning of patent claims. Additionally, because patent applications are maintained in secrecy until the application is published, we may be unaware of third-party patents that may be infringed by commercialization of our product candidate or any future product candidate. There may be certain issued patents and patent applications claiming subject matter that we may be required to license in order to research, develop or commercialize any of our product candidates, and we do not know if such patents and patent applications would be available to license on commercially reasonable terms, or at all. Any claims of patent infringement asserted by third parties would be time-consuming and may: (i) result in costly litigation; (ii) divert the time and attention of our technical personnel and management; (iii) prevent us from commercializing a product until the asserted patent expires or is held finally invalid or not infringed in a court of law; (iv) require us to cease or modify our use of the technology and/or develop non-infringing technology; or (v) require us to enter into royalty or licensing agreements.
Third parties may hold proprietary rights that could prevent our product candidate from being marketed. Any patent-related legal action against us claiming damages and seeking to enjoin commercial activities relating to any of our product candidates or our processes could subject us to potential liability for damages and require us to obtain a license to continue to manufacture or market our product candidate or any future product candidates. We cannot predict whether we would prevail in any such actions or that any license required under any of these patents would be made available on commercially acceptable terms, if at all. In addition, we cannot be sure that we could redesign our product candidate or any future product candidates or processes to avoid infringement, if necessary. Accordingly, an adverse determination in a judicial or administrative proceeding, or the failure to obtain necessary licenses, could prevent us from developing and commercializing our product candidate or a future product candidate, which could harm our business, financial condition and operating results.
48
We expect that there are other companies, including major pharmaceutical companies, working in the areas competitive to our product candidates which either has resulted, or may result, in the filing of patent applications that may be deemed related to our activities. If we were to challenge the validity of these or any issued United States patent in court, we would need to overcome a statutory presumption of validity that attaches to every issued United States patent. This means that, in order to prevail, we would have to present clear and convincing evidence as to the invalidity of the patent’s claims. If we were to challenge the validity of these or any issued United States patent in an administrative trial before the Patent Trial and Appeal Board in the U.S. PTO, we would have to prove that the claims are unpatentable by a preponderance of the evidence. There is no assurance that a jury and/or court would find in our favor on questions of infringement, validity or enforceability. Even if we are successful, litigation could result in substantial costs and be a distraction to management.
Risks Relating to Our Recycling Business
Our revenues, earnings and cash flows will fluctuate based on changes in commodity prices.
Our recycling operations process for sale certain recyclable materials, including steel, aluminium, and copper, all of which are subject to significant market price fluctuations. The majority of recyclables we process for sale are steel and non-ferrous metals. Fluctuations in market prices or demand for such commodity items, particularly from China and Turkey, can affect our future operating income and cash flows negatively. As we have increased the size of our recycling operations, we have also increased our exposure to commodity price fluctuations.
In the past we also earned a significant amount of revenue from the sale of carbon credits. The creation of carbon offsets involves a consultant's establishment of a project that includes the successful destruction of the Company's ozone-depleting refrigerants. The project process involves a significant degree of regulatory compliance and only a limited number of facilities are approved to destroy ozone-depleting refrigerants. While we no longer sell carbon credits, we currently sell our ozone-depleting refrigerants to consultants that manage the creation of carbon offsets. If we are unable to find businesses that purchase ozone-depleting refrigerants for the creation of carbon offsets or if carbon credit programs are significantly altered or discontinued, the market for these refrigerants could be reduced or eliminated. If we do not have a market for these refrigerants or if a governmental authority requires their destruction, the costs of our operations would increase, resulting in a material adverse impact on our financial condition and results of operations.
We purchase our replacement appliances from third-party manufacturers, who we believe manufacture those appliances in China and Mexico, and, as a result, international trade conditions could adversely affect us.
We purchase our replacement appliances from third-party manufacturers, who we believe manufacture certain types of those appliances in China and Mexico or purchase materials or parts from China and Mexico for use in manufacturing. As a result, tariffs, political or financial instability, labor strikes, natural disasters, public health crises (such as the coronavirus), or other events resulting in the disruption of trade or transportation from China or Mexico or the imposition of additional regulations relating to foreign trade could cause significant delays or interruptions in the supply of our merchandise or increase our costs, either of which could have an adverse effect on our business. If we were unable to adequately replace the merchandise we currently source with merchandise produced elsewhere, our business could be adversely affected.
The United States has recently imposed tariffs on various imports from China, including some of our replacement appliances. Since the imposition of these tariffs, third-party manufacturers have increased the price of the appliances we purchase from them and retain the right to implement further increases. These tariffs remain largely unmitigated and the Company cannot predict if and when the tariffs will be reduced or eliminated. The ongoing impact of these tariffs will depend on future trade discussions between the U.S. and China or the Company’s ability to avoid or offset these costs should the tariffs remain in place. The Company may not be able to pass such increased costs on to its customers, and the Company may not be able to secure sources of certain products and materials that are not subject to tariffs on a timely basis or at all. Such developments could have a material adverse impact on the Company’s business, financial condition and results of operations.
If our third-party collection or delivery services are unable to meet our promised pickup and delivery schedules, our net sales may decline due to a decline in customer satisfaction.
We offer appliance pickup and delivery services, which are significantly outsourced to third-party providers. Our third-party services are subject to risks beyond our control. If appliances are not picked up on time, or at all, or products are not delivered on time, our clients and customers may cancel their orders, or we may lose business from our clients and customers in the future. As a result, our net sales and profitability may decline.
49
Our sales may not be an indication of our future results of operations because they fluctuate significantly.
Our current and historical sales figures have fluctuated significantly from quarter to quarter. A number of factors have historically affected, and will continue to affect, our sales results and profitability, including, (i) changes in competition, such as pricing pressure; (ii) periodic sale of ozone-depleting refrigerants used in the creation of carbon offsets; (iii) fluctuating commodity prices and available markets for our byproduct sales; (iv) changes in recycling and replacement programs with utility customers; (v) weather conditions in our markets; and (vi) timing of promotional events.
Significant shortages in diesel fuel supply or increases in diesel fuel prices will increase our operating expenses.
The price and supply of diesel fuel can fluctuate significantly based on international, political and economic circumstances, as well as other factors outside our control, such as actions by the Organization of the Petroleum Exporting Countries (“OPEC”) and other oil and gas producers, regional production patterns, weather conditions and environmental concerns. Our collection and delivery agents need diesel fuel to run a significant portion of our collection and delivery of appliance activities. Supply shortages could substantially increase our operating expenses. Additionally, if fuel prices increase, our direct operating expenses will increase and many of our vendors may raise their prices as a means to offset their rising costs. We may not be able to pass through all of our increased costs to our customers and some contracts prohibit any pass-through of the increased costs.
Our revenues from recycling contracts are subject to seasonal fluctuations and are dependent on the utilities’ advertising and promotional activities for contracts in which we do not provide advertising services.
Our business is dependent largely upon our ability to obtain new contracts and continue existing contracts for appliance recycling services and appliance replacement programs with utility companies and other sponsors of energy efficiency programs. Contracts with these entities generally have initial terms of one to three years, with renewal options and early termination clauses. However, some contracts are for programs that are non-recurring. Although we continue to respond to requests for bids for upcoming recycling and replacement services, we are still dependent on certain customers for a large portion of our revenues. The loss or material reduction of business from any of these major customers could adversely affect our revenues and profitability. While we wish to add new recycling and appliance replacement contracts in 2021 and beyond, we cannot assure you that our existing contracts will continue, that they will be sufficiently profitable, that existing customers will continue to use our services at current levels or that we will be successful in obtaining new contracts going forward.
Risks Relating to Our Technology Business
GeoTraq has incurred significant operating losses since inception and expects the losses will continue into the future. If the losses continue GeoTraq may have to suspend operations or cease operations.
GeoTraq has no operating history upon which an evaluation of its future success or failure can be made. GeoTraq has incurred significant operating losses since inception and has limited financial resources to support it until such time that it is able to generate positive cash flow from operations. GeoTraq’s ability to achieve and maintain profitability and positive cash flow is dependent upon its ability to (i) develop its technology and (ii) generate revenues from its planned business operations. Based upon current plans, GeoTraq expects to continue to incur operating losses in future periods. Failure to generate revenues may cause GeoTraq to suspend or cease operations.
GeoTraq is in the early stages of development.
GeoTraq is developing a new technology and may encounter difficulties including unanticipated problems relating to the development and testing of its product, initial and continuing regulatory compliance, vendor manufacturing costs, production and assembly of its product, and the competitive and regulatory environments in which the company intends to operate. It is uncertain, at this stage of its development, if GeoTraq is unable to effectively resolve any such problems, should they occur. If GeoTraq cannot resolve an unanticipated problem, it may be forced to modify or abandon its business plan.
50
GeoTraq does not have sufficient funds to complete each phase of its proposed plan of operation and as a result may have to suspend operations.
Each of the phases of GeoTraq’s plan of operation is limited and restricted by the amount of working capital that GeoTraq has and is able to obtain from the Company, raise from financings, and generate from business operations. Initially, GeoTraq intended to finance its plan of operation with funds from the Company and private loans, and, subsequently, with revenues generated from its business operations.
Based on latest worst-case projections, GeoTraq will not generate revenues and positive cashflows from operations to satisfy its cash requirements for the next 12 months and will be required to obtain the funds from the Company or raise the required funds by way of equity or debt financing. However, these projections do expect that GeoTraq will generate sufficient revenues and cash flows from operations to satisfy its cash requirements within the next couple of years without JanOne funding assistance. Based on this, the likelihood that GeoTraq may have to suspend operations is remote.
GeoTraq outsources the research and development of its technology, and as a result it is dependent upon those third-party developers to develop our products in a timely and cost-efficient manner while maintaining a minimum level of quality.
GeoTraq does not have internal manufacturing capabilities and relies on contract manufacturers to manufacture and develop its products. GeoTraq cannot be certain that it will not experience operational difficulties with its future manufacturers, including reductions in the availability of production capacity, errors in complying with product specifications, insufficient quality control, failures to meet production deadlines, increases in manufacturing costs and increased lead times. Additionally, GeoTraq’s future manufacturers may experience disruptions in their manufacturing operations due to equipment breakdowns, labor strikes or shortages, component or material shortages, cost increases or other similar problems. Further, in order to minimize their inventory risk, GeoTraq’s future manufacturers might not order components from third-party suppliers with adequate lead time, thereby impacting its ability to meet demand forecasts. Therefore, if GeoTraq fails to manage its relationship with its manufacturers effectively, or if they experience operational difficulties, GeoTraq’s ability to ship products could be impaired and its competitive position and reputation could be harmed.
In the event that GeoTraq receives shipments of products that fail to comply with its technical specifications or that fail to conform to its quality control standards, and it is not able to obtain replacement products in a timely manner, GeoTraq risks revenue losses from the inability to sell those products, increased administrative and shipping costs, and lower profitability. Additionally, if defects are not discovered until after customers purchase its products, GeoTraq customers could lose confidence in the technical attributes of its products and its business could be harmed.
GeoTraq will not control its future contract manufacturers or suppliers, including their labor, environmental or other practices, or require them to comply with a formal code of conduct. However, GeoTraq intends to conduct periodic audits of its contract manufacturers’ and suppliers’ compliance with applicable laws and good industry practices, these audits may not be frequent or thorough enough to detect non-compliance. A violation of labor, environmental or other laws by its contract manufacturers or suppliers, or a failure of these parties to follow ethical business practices, could lead to negative publicity and harm GeoTraq’s reputation. In addition, GeoTraq may choose to seek alternative manufacturers or suppliers if these violations or failures were to occur. Identifying and qualifying new manufacturers or suppliers can be time consuming and GeoTraq may not be able to substitute suitable alternatives in a timely manner or at an acceptable cost. Other consumer products companies have faced significant criticism for the actions of their manufacturers and suppliers, and GeoTraq could face such criticism as well. Any of these events could adversely affect its brand, harm its reputation, reduce demand for its products and harm its ability to meet demand if it needs to identify alternative manufacturers or suppliers.
51
Cellular service providers on which GeoTraq’s technology is dependent may change the terms by which the technology is used on their networks, which could result in lower revenue and adverse effects on our business.
If the cellular service providers on which GeoTraq’s technology is to be used changes the terms of use or eliminates the ability to use products that incorporate GeoTraq’s technology, GeoTraq could lose customers as they would no longer be able to use GeoTraq’s technology in their products. In addition, GeoTraq could be required to change its fee structure to retain customers, which could negatively affect GeoTraq’s gross margins. The cellular service providers may also decide to raise prices, impose usage caps or fees, or discontinue certain application bundles, which could adversely affect end users who use GeoTraq’s technology. If imposed, these pricing changes or usage restrictions could make GeoTraq’s technology less attractive and could result in current end users abandoning GeoTraq’s technology. If end user turnover increased, the number of GeoTraq’s end users and GeoTraq’s revenue would decrease and its business would be harmed.
GeoTraq’s ability to increase or maintain its customer base and revenue will be impaired if cellular service providers do not allow GeoTraq Technology access to their networks.
GeoTraq’s technology requires cellular service to operate. The products produced by manufactures will require end users to maintain service with cellular service providers. If cellular service providers do not permit end users to purchase the cellular connectivity the product requires, GeoTraq may have difficulty attracting manufacturing customers because of the lack of, or difficulty in purchasing and provisioning a service plan. If the end user is unable to provide seamless provisioning or the carrier cancels their subscriptions, GeoTraq’s business may be harmed.
GENERAL RISK FACTORS
Isaac Capital Group, LLC (“ICG”) owns a large percentage of our voting stock, which may allow them to control substantially all matters requiring stockholder approval.
Currently, ICG owns approximately 16.3% of our outstanding shares of common stock. ICG’s sole member is Jon Isaac, the President and Chief Executive Officer of Live Ventures Incorporated. Jon Isaac is the son of our Chief Executive Officer Tony Isaac. Because of such ownership and the relationship, ICG may be able to significantly, and possibly adversely, affect our corporate decisions, including the election of the board of directors.
The market price of our common stock has been, and may continue to be volatile and fluctuate significantly, which could result in substantial losses for investors and subject us to securities class action litigation.
The trading price for our common stock has been, and we expect it to continue to be, volatile. The price at which our common stock trades depends upon a number of factors, including our historical and anticipated operating results, our financial situation, announcements of technological innovations or new products by us, our ability or inability to raise the additional capital we may need and the terms on which we raise it, and general market and economic conditions. Some of these factors are beyond our control. Broad market fluctuations may lower the market price of our common stock and affect the volume of trading in our stock, regardless of our financial condition, results of operations, business or prospect. Among the factors that may cause the market price of our common stock to fluctuate are the risks described in this “Risk Factors” section. In addition, the stock markets, in general, The Nasdaq Capital Market and the market for biopharmaceutical companies in particular, may experience a loss of investor confidence. Such loss of investor confidence may result in extreme price and volume fluctuations in our common stock that are unrelated or disproportionate to the operating performance of our business, financial condition or results of operations. These broad market and industry factors may materially harm the market price of our common stock and expose us to securities class action litigation. Such litigation, even if unsuccessful, could be costly to defend and divert management’s attention and resources, which could further materially harm our financial condition and results of operations.
52
Our executive offices are located in Las Vegas, Nevada in a leased facility consisting of 11,000 square feet of office space.
Recycling Centers
We lease the recycling center facilities described below.
|
Approximate Sqft |
|
Location |
|
5,000 |
|
Dartmouth, Nova Scotia |
|
18,500 |
|
Santa Fe Springs, California |
|
5,900 |
|
Albuquerque, New Mexico |
|
14,600 |
|
Minneapolis, Minnesota |
|
12,000 |
|
Indianapolis, Indiana |
|
19,800 |
|
Franklin, Massachusetts |
|
7,500 |
|
Commerce City, Colorado |
|
12,100 |
|
Cudahy, Wisconsin |
|
23,200 |
|
Pittsburgh, Pennsylvania |
|
14,300 |
|
Mechanicsburg, Pennsylvania |
|
38,000 |
|
Philadelphia, Pennsylvania |
|
30,000 |
|
Syracuse, New York |
|
12,800 |
|
Sacramento, California |
|
14,600 |
|
Norcross, Georgia |
|
7,400 |
|
North Haven, Connecticut |
|
11,700 |
|
Jackson, Mississippi |
|
3,000 |
|
Baltimore, Maryland |
|
19,200 |
|
Grand Rapids, Michigan |
The information in response to this item is included in Note 15, Commitments and Contingencies, to the Consolidated Financial Statements included in Part II, Item 8, of this Form 10-K.
ITEM 4. MINE SAFETY DISCLOSURES
None.
53
ITEM 5. MARKET FOR OUR COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information and Dividends
Our common stock trades under the symbol “JAN” on the NASDAQ Capital Market. As of March 25, 2021, there were 31 stockholders of record, which excludes stockholders whose shares were held in nominee or street name by brokers. We have no record of the number of holders of our common stock who hold their shares in “street name” with various brokers.
We have not paid dividends on our common stock and do not presently plan to pay dividends on our common stock for the foreseeable future.
Information concerning securities authorized for issuance under equity compensation plans is included in Part III, Item 12 of this report.
ITEM 6. SELECTED FINANCIAL DATA
Not applicable.
54
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
For a description of our significant accounting policies and an understanding of the significant factors that influenced our performance during the year ended January 2, 2021, this “Management’s Discussion and Analysis of Financial Condition and Results of Operations” (hereafter referred to as “MD&A”) should be read in conjunction with the consolidated financial statements, including the related notes, appearing in Part II, Item 8 of this Annual Report on Form 10-K (this “Form 10-K”) for the fiscal year ended January 2, 2021.
Amounts are stated in thousands of dollars expect share and per share amounts.
Note about Forward-Looking Statements
This Form 10-K includes statements that constitute “forward-looking statements.” These forward-looking statements are often characterized by the terms “may,” “believes,” “projects,” “intends,” “plans,” “expects,” or “anticipates,” and do not reflect historical facts. Specific forward-looking statements contained in this portion of the Form 10-K include, but are not limited to: (i) statements relating to our initial product candidate, JAN101, including statements relating to the commencement of Phase 2b clinical trials for the treatment of PAD in 2021 and the results of those trials, (ii) statements that are based on current projections and expectations about the markets in which we operate, (iii) statements relating to the sale of the Company’s Recycling business, (iv) statements about current projections and expectations of general economic conditions, (v) statements about specific industry projections and expectations of economic activity, (vi) statements relating to our future operations and prospects, (vii) statements about future results and future performance, (viii) statements that the cash on hand and additional cash generated from operations together with potential sources of cash through issuance of debt or equity will provide the Company with sufficient liquidity for the next 12 months, and (ix) statements that the outcome of pending legal proceedings will not have a material adverse effect on business, financial position and results of operations, cash flow or liquidity.
Forward-looking statements involve risks, uncertainties and other factors, which may cause our actual results, performance or achievements to be materially different from those expressed or implied by such forward-looking statements. Factors and risks that could affect our results, future performance and capital requirements and cause them to materially differ from those contained in the forward-looking statements include those identified in this Form 10-K under Item 1A “Risk Factors”, as well as other factors that we are currently unable to identify or quantify, but that may exist in the future.
In addition, the foregoing factors may generally affect our business, results of operations and financial position. Forward-looking statements speak only as of the date the statements were made. We do not undertake and specifically decline any obligation to update any forward-looking statements. Any information contained on our website www.janone.com or any other websites referenced in this Form 10-K are not part of this Form 10-K.
Our Company
JanOne is focused on finding treatments for conditions that cause severe pain and bringing to market drugs with non-addictive pain-relieving properties. In addition, through our subsidiaries ARCA Recycling, Inc., Customer Connexx LLC, and ARCA Canada Inc., JanOne is engaged in the business of recycling major household appliances in North America by providing turnkey appliance recycling and replacement services for utilities and other sponsors of energy efficiency programs. In addition, through its GeoTraq Inc. (“GeoTraq”) subsidiary, we are engaged in the development, design and, ultimately, we expect the sale of wireless transceiver modules with technology that provides LBS directly from global Mobile IoT networks
We operate three reportable segments:
|
|
• |
Biotechnology: Our biotechnology segment is focused on finding treatments for conditions that cause severe pain and bringing to market drugs with non-addictive pain-relieving properties. |
|
|
• |
Recycling: Our recycling segment is a turnkey appliance recycling program. We receive fees charged for recycling, replacement and additional services for utility energy efficiency programs and have established 18 Regional Processing Centers (“RPCs”) for this segment throughout the United States and Canada |
55
|
|
• |
Technology: GeoTraq is in the process of developing technology to enable low cost, location-based products and services. |
Reporting Period. We report on a 52-or 53-week fiscal year. Our 2020 fiscal year ended on January 2, 2021 (“fiscal 2020”). Our 2019 fiscal year ended on December 28, 2019 (“fiscal 2019”).
Application of Critical Accounting Policies
Our discussion of the financial condition and results of operations is based upon our consolidated financial statements, which have been prepared in conformity with accounting principles generally accepted in the United States. The preparation of our consolidated financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, revenues and expenses, and related disclosure of any contingent assets and liabilities at the date of the financial statements. Management regularly reviews its estimates and assumptions, which are based on historical factors and other factors believed to be relevant under the circumstances. Actual results may differ from these estimates under different assumptions, estimates or conditions.
Critical accounting policies are defined as those that are reflective of significant judgments and uncertainties and potentially result in materially different results under different assumptions and conditions. ARCA’s critical accounting policies include intangible impairment under ASC 350, revenue recognition under ASC 606, and going concern under ASC 205.
Results of Operations
The following table sets forth certain statement of operations items from continuing operations and as a percentage of revenue, for the periods indicated:
|
|
|
Fiscal Year Ended |
|
|
Fiscal Year Ended |
|
||||||||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||||||||||
|
Statement of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues |
|
$ |
33,867 |
|
|
|
100.0 |
% |
|
$ |
35,097 |
|
|
|
100.0 |
% |
|
Cost of revenues |
|
|
25,040 |
|
|
|
73.9 |
% |
|
|
27,311 |
|
|
|
77.8 |
% |
|
Gross profit |
|
|
8,827 |
|
|
|
26.1 |
% |
|
|
7,786 |
|
|
|
22.2 |
% |
|
Selling, general and administrative expenses |
|
|
17,823 |
|
|
|
52.6 |
% |
|
|
20,217 |
|
|
|
57.6 |
% |
|
Operating loss |
|
|
(8,996 |
) |
|
|
(26.6 |
)% |
|
|
(12,431 |
) |
|
|
(35.4 |
)% |
|
Interest expense, net |
|
|
(504 |
) |
|
|
(1.5 |
)% |
|
|
(1,480 |
) |
|
|
(4.2 |
)% |
|
Impairment charges |
|
|
— |
|
|
|
— |
|
|
|
(2,992 |
) |
|
|
(11.0 |
)% |
|
Gain on litigation settlement |
|
|
418 |
|
|
|
4.7 |
% |
|
|
694 |
|
|
|
8.9 |
% |
|
Gain on settlement of vendor advance payments |
|
|
142 |
|
|
|
0.8 |
% |
|
|
— |
|
|
|
— |
|
|
Other income |
|
|
15 |
|
|
|
0.0 |
% |
|
|
1,048 |
|
|
|
3.0 |
% |
|
Net loss before income taxes |
|
|
(8,925 |
) |
|
|
(26.4 |
)% |
|
|
(15,161 |
) |
|
|
(43.2 |
)% |
|
Benefit from income taxes |
|
|
427 |
|
|
|
1.3 |
% |
|
|
3,197 |
|
|
|
9.1 |
% |
|
Net loss |
|
$ |
(8,498 |
) |
|
|
(25.1 |
)% |
|
$ |
(11,964 |
) |
|
|
(34.1 |
)% |
56
The following tables set forth revenues for key product and service categories, percentages of total revenue and gross profits earned by key product and service categories and gross profit percent as compared to revenues for each key product category indicated:
|
|
|
Fiscal Year Ended |
|
|
Fiscal Year Ended |
|
||||||||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||||||||||
|
|
|
Net |
|
|
Percent |
|
|
Net |
|
|
Percent |
|
||||
|
|
|
Revenue |
|
|
of Total |
|
|
Revenue |
|
|
of Total |
|
||||
|
Revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Recycling and Byproducts |
|
$ |
18,262 |
|
|
|
53.9 |
% |
|
$ |
21,445 |
|
|
|
61.1 |
% |
|
Replacement Appliances |
|
|
15,605 |
|
|
|
46.1 |
% |
|
|
13,652 |
|
|
|
38.9 |
% |
|
Total Revenue |
|
$ |
33,867 |
|
|
|
100.0 |
% |
|
$ |
35,097 |
|
|
|
100.0 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fiscal Year Ended |
|
|
Fiscal Year Ended |
|
||||||||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||||||||||
|
|
|
Gross |
|
|
Gross |
|
|
Gross |
|
|
Gross |
|
||||
|
|
|
Profit |
|
|
Profit % |
|
|
Profit |
|
|
Profit % |
|
||||
|
Gross Profit |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Recycling and Byproducts |
|
$ |
2,005 |
|
|
|
11.0 |
% |
|
$ |
3,890 |
|
|
|
18.1 |
% |
|
Replacement Appliances |
|
|
6,822 |
|
|
|
43.7 |
% |
|
|
3,896 |
|
|
|
28.5 |
% |
|
Total Gross Profit |
|
$ |
8,827 |
|
|
|
26.1 |
% |
|
$ |
7,786 |
|
|
|
22.2 |
% |
Revenue
Revenue remained relatively constant for the fiscal year ended January 2, 2021 as compared to the fiscal year ended December 28, 2019. Replacement Appliance revenue increased $1,953 or 14.3% due to higher volumes from two customers, offset by a decrease in Recycling and Byproducts revenue of $3,183 or 14.8% due to the temporary closure of our facilities as a result of COVID-19 and decreased volumes from our customers.
Cost of Revenue
Cost of revenue decreased $2,271, or 8.3% for the fiscal year ended January 2, 2021 as compared to the fiscal year ended December 28, 2019, primarily due to cost cutting efforts and the temporary closure of our facilities due to COVID-19.
Selling, General and Administrative Expense
Selling, general and administrative expense decreased $2,394 or 11.8%, for the fiscal year ended January 2, 2021 as compared to the fiscal year ended December 28, 2019, primarily due to a temporary reduction in employees, cost cutting measures and other activities related to the closure of our facilities due to COVID-19.
Interest Expense, net
Interest expense net decreased $976 or 65.9%, for the fiscal year ended January 2, 2021 as compared to the fiscal year ended December 28, 2019 primarily due to the decrease in the related party debt balance.
Impairment Charges
On December 9, 2019, ApplianceSmart, Inc. (“ApplianceSmart”), a former subsidiary of the Company, filed a voluntary petition in the United States Bankruptcy Court for the Southern District of New York seeking relief under Chapter 11 of Title 11 of the United States Code. As a result, the Company has recorded an impairment charge of $2,992 for the amount owed by ApplianceSmart to the Company as of December 28, 2019. There were no similar impairment charges for the fiscal year ended January 2, 2021.
57
See Note 4 of the Consolidated Financial Statements for a complete discussion of the ApplianceSmart Note.
Gain (Loss) on Litigation Settlement
As of January 2, 2021, the Company recorded a net gain on litigation settlement of $418 comprised of the $800 gain on settlement of litigation with a former service provider (discussed below), partially offset by a loss on settlement of outstanding payables of $382.
On October 4, 2018, the Company initiated litigation against a former professional services provider (“PSP”), in Illinois state court, as well as a private arbitration proceeding that was scheduled to be held in Minneapolis, Minnesota, arising from PSP’s rendering of certain professional services to the Company during the period from 2011 through 2014. PSP filed a counterclaim in the arbitration seeking an award of its legal fees and costs arising from that proceeding. The parties subsequently agreed to consolidate their respective claims into the arbitration. The Company’s arbitration demand, as amended, sought an award of more than $50 and other relief. On March 23, 2020, the parties entered into a settlement agreement, whereby, without any admission of liability, they exchanged mutual releases, agreed to dismiss their respective claims with prejudice, and PSP agreed to pay $800 to the Company to, among other things, assist it with certain of its costs and obligations that related to various issues underlying the arbitration proceeding.
On November 15, 2016, the Company served an arbitration demand on Haier US Appliance Solutions, Inc., dba GE Appliances (“GEA”), alleging breach of contract and interference with prospective business advantage. On April 18, 2017, GEA served a counterclaim regarding alleged obligations under the parties’ recycling agreement. On December 12, 2017, the court stayed GEA’s complaint in favor of the arbitration. Under the terms of the Company’s transaction with Recleim LLC (“Recleim”), Recleim is obligated to pay GEA on the Company’s behalf the amounts claimed by GEA in the arbitration and in the lawsuit pending in Kentucky. Those amounts have been paid into escrow pending the outcome of the arbitration. Arbitration proceedings were held in October and November 2019. On March 5, 2020, the arbitrator ruled in part in favor of the Company and in part in favor of GEA, and, as a result, the Company recorded a gain on litigation settlement of $694.
See Note 15 of the Consolidated Financial Statements for a complete litigation discussion.
Other Income
Other income was $15 for the fiscal year ended January 2, 2021 as compared to $1,048 the fiscal year ended December 28, 2019 primarily due to the discussion below.
Sears Holdings Management Corp – Logistics Services
On February 18, 2019, the Company informed Sears Holdings Management Corp – Logistics Services (“Sears”) that Sears may have overcharged ARCA Recycling $642 and that it planned on filing a proof of claim with the trustee in the Sears’ bankruptcy against Sears for the overcharged amount. The Company requested that Sears provide contractual written proof to the contrary supporting their claim for invoices submitted in excess of the contractually agreed upon amounts for transportation services. Sears provided transportation services to ARCA Recycling in fiscal years 2013 through 2018. ARCA Recycling recorded $559 as outstanding and un-paid accounts payable as of December 28, 2019. The Company wrote off the overcharged amount of $559 as other income in the consolidated results for the fiscal year ended December 28, 2019.
Segment Performance
We report our business in the following segments: Biotechnology, Recycling and Technology. We identified these segments based on a combination of business type, customers serviced and how we divide management responsibility. Our revenues and profits are driven through our recycling centers, e-commerce, individual sales reps and our internet services for our recycling and technology segment. We expect revenues and profits for our biotechnology segment to be driven by the development of pharmaceuticals that treat the root cause of pain but are non-opioid painkillers. We include Corporate expenses within the Recycling segment.
58
Operating loss by operating segment, is defined as loss before net interest expense, other income and expense, provision for income taxes.
|
|
|
Fiscal Year Ended January 2, 2021 |
|
|
Fiscal Year Ended December 28, 2019 |
|
||||||||||||||||||||||||||
|
|
|
Biotechnology |
|
|
Recycling |
|
|
Technology |
|
|
Total |
|
|
Biotechnology |
|
|
Recycling |
|
|
Technology |
|
|
Total |
|
||||||||
|
Revenue |
|
$ |
— |
|
|
$ |
33,867 |
|
|
$ |
— |
|
|
$ |
33,867 |
|
|
$ |
— |
|
|
$ |
35,097 |
|
|
$ |
— |
|
|
$ |
35,097 |
|
|
Cost of revenue |
|
|
— |
|
|
|
25,040 |
|
|
|
— |
|
|
|
25,040 |
|
|
|
— |
|
|
|
27,311 |
|
|
|
— |
|
|
|
27,311 |
|
|
Gross profit |
|
|
— |
|
|
|
8,827 |
|
|
|
— |
|
|
|
8,827 |
|
|
|
— |
|
|
|
7,786 |
|
|
|
— |
|
|
|
7,786 |
|
|
Selling, general and administrative expense |
|
|
1,738 |
|
|
|
11,999 |
|
|
|
4,086 |
|
|
|
17,823 |
|
|
|
1,038 |
|
|
|
14,183 |
|
|
|
4,996 |
|
|
|
20,217 |
|
|
Operating loss |
|
$ |
(1,738 |
) |
|
$ |
(3,172 |
) |
|
$ |
(4,086 |
) |
|
$ |
(8,996 |
) |
|
$ |
(1,038 |
) |
|
$ |
(6,397 |
) |
|
$ |
(4,996 |
) |
|
$ |
(12,431 |
) |
Biotechnology Segment
Our biotechnology segment commenced operations in September 2019, and, as a result, incurred expenses of $1,038 and $1,738 related to employee costs and the operating license issued during the fourth quarter of 2019 and fiscal 2020, respectively.
Recycling Segment
The recycling segment consists of ARCA Recycling, Customer Connexx, and ARCA Canada. Revenue remained relatively flat for the fiscal year ended January 2, 2021 as compared to the fiscal year ended December 28, 2019. Replacement Appliance revenue increased $1,953 or 14.3% due to higher volumes from two customers, offset by a decrease in Recycling and Byproducts revenue of $3,183 or 14.8% due to the temporary closure of our facilities as a result of COVID-19 and decreased volumes from our customers.
Cost of revenue decreased $2,271, or 8.3% for the fiscal year ended January 2, 2021 as compared to the fiscal year ended December 28, 2019, primarily due to cost cutting efforts and the temporary closure of our facilities due to COVID-19.
Operating loss for the fiscal year ended January 2, 2021, decreased $3,225 or 50.4% as compared to the prior year period. This represents an increase in gross profit of $1,041 and a decrease in selling, general and administrative expense of $2,184. The decrease in selling, general and administrative expense is primarily attributable to due to a temporary reduction in employees, cost cutting measures and other activities related to the closure of our facilities due to COVID-19.
Technology Segment
The technology segment consists of GeoTraq. Results for the fiscal year ended January 2, 2021 include a loss of $4,086 which approximated the fiscal year ended December 28, 2019 loss of $4,996. The loss represents intangible asset amortization expense and other selling general and administrative expense for each period.
Liquidity and Capital Resources
Overview
As of January 2, 2021, we had total cash on hand of $379. As we continue to prepare to begin late-stage clinical development with our pharmaceutical product, JAN101, and potentially pursue strategic transactions to expand and grow our business, we regularly monitor capital market conditions and may raise additional funds through borrowings or public or private sales of debt or equity securities. The amount, nature and timing of any borrowings or sales of debt or equity securities will depend on our operating performance and other circumstances; our then-current commitments and obligations; the amount, nature and timing of our capital requirements; any limitations imposed by our current credit arrangements; and overall market conditions.
59
In December 2019, the 2019 novel coronavirus (COVID-19) surfaced in Wuhan, China. The World Health Organization declared a global emergency on January 30, 2020, and most countries initiated travel restrictions limiting travel to other countries and lock-downs within their borders. While various vaccines have recently been introduced into the marketplace, the impacts of variant strains of the COVID-19 virus is still unknown. The widespread health crisis has adversely affected the global economy, resulting in an economic downturn that could impact demand for our products. To date, the outbreak had a material adverse impact on our operations. For example, several customers in our appliance recycling and appliance replacement business have previously suspended our ability to pick up and or replace their customers’ appliances resulting in decreased revenues for both recycling and replacement business. The future impact of the outbreak is highly uncertain and cannot be predicted and there is no assurance that the outbreak will not have another material adverse impact on the future results of the Company. The extent of the impact, if any, will depend on future developments, including actions taken to contain the coronavirus. A key task for the Company in 2021 is to begin late-stage clinical development with its pharmaceutical product, JAN101. However, the COVID-19 pandemic significantly impacted clinical trials in 2020, delaying recruitment in most non-COVID-19 clinical trials and even eliminating recruitment in some trials. While clinical sites have largely resumed conducting non-COVID-19 clinical trials, the backlog of subjects may adversely affect our ability to recruit for its trial, leading to longer and more expensive trials. In addition, the unknown effectiveness of the COVID-19 vaccines, particularly concerning variant strains of COVID-19, could lead to clinical sites terminating patient recruitment again during the course of the study.
On May 1, 2020, the Company entered into a promissory note (the “Promissory Note”) with Texas Capital Bank, N.A. that provides for a loan in the amount of $1,872 (the “PPP Loan”) pursuant to the Paycheck Protection Program under the Coronavirus Aid, Relief and Economic Security Act (the “CARES Act”). The PPP Loan matures on April 27, 2022 and bears interest at a rate of 1.0% per annum. Monthly amortized principal and interest payments are deferred for six months after the date of disbursement. The Promissory Note contains events of default and other provisions customary for a loan of this type. The Paycheck Protection Program provides that the use of PPP Loan amount shall be limited to certain qualifying expenses and may be partially or wholly forgiven in accordance with the requirements set forth in the CARES Act. The Company has applied for forgiveness of a portion of the loan in accordance with the terms of the CARES Act to the extent applicable.
As of the period ending September 26, 2020, the Company received advance payments authorized by the California Public Utilities Commission and processed through two California utilities for the purposes of sustaining the workforce during the COVID 19 pandemic shutdown. The use of these funds was limited to labor and labor benefits for impacted employees. Portions of these advances are forgivable if certain conditions are met the specifics which have not been finalized. Advance payments that are not forgiven will need to be paid back in full by December 31, 2021. Total funding received under this program as of September 26, 2020 amounted to $1,168. As of January 2, 2021, $142 was forgiven, leaving a balance remaining of $1,026.
On January 29, 2021, the Company entered into a Securities Purchase Agreement (the “Purchase Agreement”) with certain institutional investors (the “Purchasers”) for the sale by the Company in a registered direct offering (the “Offering”) of 571,428 shares of the Company’s common stock, par value $0.001 per share (the “Common Stock”), at a purchase price per share of Common Stock of $10.50.
On February 2, 2021, the Offering closed and the Company received gross proceeds of approximately $6,000, before deducting placement agent fees and other offering expenses. The Company is utilizing the net proceeds for general working capital.
Based on our current operating plans, we believe that available cash balances, funds available under our factoring agreement with Prestige Capital Finance, LLC (“Prestige Capital”), and or other refinancing of existing indebtedness will provide sufficient liquidity to fund our operations, our continued investments in store openings and remodeling activities for at least the next 12 months.
60
Cash Flows
During the fiscal year ended January 2, 2021, cash used in operations was $617, compared to cash used in operations of $3,510 during the fiscal year ended December 28, 2019. The decrease in cash used in operations was primarily due to the decrease in net loss, discussed above, offset by noncash decrease impairment charges of $2,992 and a decrease in deferred income taxes of $3,009. Additionally, changes in working capital accounts affecting operating cash flows were as follows: a decrease in accounts receivable of $3,745 and accounts payable and accrued expenses of $3,023.
Cash used in investing activities was $834 for the fiscal year ended January 2, 2021 was attributable to purchases of property and equipment and intangibles. Cash provided by investing activities of $345 for fiscal year ended December 28, 2019 was primarily attributable the net payments received on a note receivable from ApplianceSmart of $845, offset by the purchases of property and equipment of $212 and intangible assets of $288.
Cash provided by financing activities was $1,404 for the fiscal year ended January 2, 2021 was related to proceeds from short term debt of $3,469 primarily associated with the Payroll Protection Program and advances from certain customers for future services and payment of $1,500 on its related party note. Cash provided by financing activities was $2,462 for the fiscal year ended December 28, 2019 was primarily related to the $2,500 proceeds on the related party note.
Sources of Liquidity
We utilize cash on hand and factor on occasion certain accounts receivable invoices to cover normal and seasonal fluctuations in cash flows and to support our various growth initiatives. Our cash and cash equivalents are carried at cost and consist primarily of demand deposits with commercial banks. On March 26, 2018, the Company entered into a purchase and sale agreement with Prestige Capital, whereby from time to time the Company can factor certain accounts receivable to Prestige Capital up to a maximum advance and outstanding balance of $11,000. Discount fees ultimately paid depend upon how long an invoice and related amount is outstanding from ARCA Recycling’s customer. Prestige Capital has been granted a security interest in all ARCA Recycling’s accounts receivable. The current purchase and sale agreement with Prestige Capital automatically renews every six months unless terminated by the parties.
We acknowledge that we continue to face a challenging competitive environment as we continue to focus on our overall profitability, including managing expenses. We reported a net loss of $8,498 and $11,964 in 2020 and 2019, respectively. In addition, the Company has total current assets of $6,941 and total current liabilities $20,597 resulting in a net negative working capital of $13,656.
In Item 1A. Risk Factors, management has addressed and evaluated the risk factors that could materially and adversely affect the entity’s business, financial condition and results of operations, cash flows and liquidity. The Company has determined the risk factors do not materially affect the Company’s ability to continue as a going concern within one year after the date that the financial statements are issued.
Based on the above, management has concluded that the Company is not aware and did not identify any other conditions or events that would cause the Company to not be able to continue business as a going concern for the next twelve months.
Future Sources of Cash; New Acquisitions, Products and Services
We may require additional debt financing and/or capital to finance new acquisitions, refinance existing indebtedness or consummate other strategic investments in our business. Any financing obtained may further dilute or otherwise impair the ownership interest of our existing stockholders.
Off Balance Sheet Arrangements
At January 2, 2021, we had no off-balance sheet arrangements, commitments or guarantees that require additional disclosure or measurement.
61
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Market Risk and Impact of Inflation
Interest Rate Risk. We do not believe there is any significant risk related to interest rate fluctuations on our short and long-term fixed rate debt.
Foreign Currency Exchange Rate Risk. We currently generate revenues in Canada. The reporting currency for our consolidated financial statements is U.S. dollars. It is not possible to determine the exact impact of foreign currency exchange rate changes; however, the effect on reported revenue and net earnings can be estimated. We estimate that the overall strength of the U.S. dollar against the Canadian dollar had an immaterial impact on the revenues and net income for the fiscal year ended January 2, 2021. We do not currently hedge foreign currency fluctuations and do not intend to do so for the foreseeable future.
We do not hold any derivative financial instruments, nor do we hold any securities for trading or speculative purposes.
62
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
|
Description |
|
Page |
|
|
|
|
|
|
F-1 |
|
|
|
|
|
|
Consolidated Balance Sheets as of January 2, 2021 and December 28, 2019 |
|
F-4 |
|
|
|
|
|
|
F-5 |
|
|
|
|
|
|
|
F-6 |
|
|
|
|
|
|
|
F-7 |
|
|
|
|
|
|
|
F-8 |
63
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of JanOne Inc.
Las Vegas, Nevada
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of JanOne Inc. (the “Company”) as of January 2, 2021 and December 28, 2019, and the related consolidated statements of operations and comprehensive loss, changes in stockholders’ equity, and cash flows for each of the two years in the period ended January 2, 2021, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at January 2, 2021 and December 28, 2019, and the results of their operations and their cash flows for each of the two years in the period ended January 2, 2021, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
F-1
Revenue Recognition
As described in Notes 2 and 21 to the consolidated financial statements, the Company’s consolidated revenue balance was $33.8 million for the year ended January 2, 2021. The Company recognizes revenue at the point in time when control is transferred to the end user, when the Company’s performance obligations are satisfied, which typically occurs upon delivery from the Company’s center facility and installation at the end user’s home.
We identified revenue recognition as a critical audit matter. Auditing revenue recognition involved especially challenging, subjective or complex auditor judgment due to the nature and extent of audit effort required to address this matter.
The primary procedures we performed to address this critical audit matter included:
|
|
• |
Evaluating the Company’s revenue recognition policy for conformity with accounting principles generally accepted in the United States of America. |
|
|
• |
Inspected executed contracts to identify the relevant performance obligations and evaluated the accounting treatment for each performance obligation. |
|
|
• |
Testing individual revenue transactions for proper revenue recognition in accordance with the Company’s revenue recognition policy. |
|
|
• |
Assessing the Company’s disclosures related to revenue recognition for conformity with accounting principles generally accepted in the United States of America. |
Intangible Asset Impairment Assessment
As described in Notes 2 and 8 to the consolidated financial statements, the Company’s intangible assets balance was $14 million as of January 2, 2021. Intangible assets are tested for recoverability annually or whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. The determination of the recoverability of intangible assets requires management to make significant estimates and assumptions related to forecasts of expected cash flows to be derived from the asset group for the remaining useful life of the asset group’s primary asset.
We identified the intangible asset impairment assessment as a critical audit matter based on the materiality of the amounts involved together with the inherent subjectivity related to the principal assumptions, which are dependent on current and future economic factors, including uncertainties arising from the COVID-19 pandemic; hence the assessment of the carrying value of intangible assets is considered to be complex and determined to be a critical audit matter in our current period audit. Auditing management’s judgments regarding forecasts of future cash flows from the intangible assets involved a high degree of subjectivity.
The primary procedures we performed to address this critical audit matter included:
|
|
• |
Obtaining an understanding of management’s process for its impairment assessment and analysis for the intangible assets. |
|
|
• |
Evaluating management’s ability to accurately forecast by comparing actual and budgeted results with historical performance, understanding changes to the Company’s business model, and determining whether assumptions used in the forecast were consistent with evidence obtained in other areas of the audit. |
|
|
• |
Evaluated the appropriateness of judgments applied by management while assessing the future potential impact of the COVID-19 pandemic. |
|
|
• |
Obtaining third-party evidence in support of management’s assumptions used in developing the forecasted cash flows from the intangible assets. |
F-2
|
|
• |
Performing independent sensitivity analysis of key assumptions, including the timing and amount of forecasted cash flows to assess the effect of possible variations on the current estimated value for the intangible assets. |
/s/ WSRP LLP
We have served as the Company's auditor since 2019.
Salt Lake City, Utah
March 30, 2021
F-3
JANONE INC.
(Dollars in thousands, except per share amounts)
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
379 |
|
|
$ |
481 |
|
|
Trade and other receivables, net |
|
|
3,600 |
|
|
|
6,578 |
|
|
Income taxes receivable |
|
|
196 |
|
|
|
76 |
|
|
Inventories |
|
|
1,630 |
|
|
|
1,348 |
|
|
Prepaid expenses and other current assets |
|
|
1,136 |
|
|
|
356 |
|
|
Total current assets |
|
|
6,941 |
|
|
|
8,839 |
|
|
Property and equipment, net |
|
|
732 |
|
|
|
324 |
|
|
Right of use asset - operating leases |
|
|
2,458 |
|
|
|
1,894 |
|
|
Intangible assets, net |
|
|
13,989 |
|
|
|
17,705 |
|
|
Deposits and other assets |
|
|
231 |
|
|
|
272 |
|
|
Total assets |
|
$ |
24,351 |
|
|
$ |
29,034 |
|
|
Liabilities and Stockholders' Equity |
|
|
|
|
|
|
|
|
|
Liabilities: |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
4,701 |
|
|
$ |
4,365 |
|
|
Accrued liabilities - other |
|
|
4,888 |
|
|
|
3,938 |
|
|
Accrued liability - California Sales Taxes |
|
|
5,769 |
|
|
|
5,438 |
|
|
Lease obligation short term - operating leases |
|
|
1,197 |
|
|
|
1,079 |
|
|
Short term debt |
|
|
3,042 |
|
|
|
280 |
|
|
Related party note |
|
|
1,000 |
|
|
|
2,473 |
|
|
Total current liabilities |
|
|
20,597 |
|
|
|
17,573 |
|
|
Lease obligation long term - operating leases |
|
|
1,388 |
|
|
|
850 |
|
|
Deferred income taxes, net |
|
|
— |
|
|
|
270 |
|
|
Total liabilities |
|
|
21,985 |
|
|
|
18,693 |
|
|
Commitments and Contingencies (Note 15) |
|
|
|
|
|
|
|
|
|
Stockholders' equity: |
|
|
|
|
|
|
|
|
|
Preferred stock, series A-1 - par value $0.001 per share 2,000,000 authorized, 259,729 and 259,729 shares issued and outstanding at January 2, 2021 and December 28, 2019, respectively |
|
|
— |
|
|
|
— |
|
|
Common stock, par value $0.001 per share, 200,000,000 shares authorized, 1,829,982 and 1,919,048 shares issued and outstanding at January 2, 2021 and at December 28, 2019, respectively |
|
|
2 |
|
|
|
2 |
|
|
Additional paid in capital |
|
|
39,869 |
|
|
|
39,291 |
|
|
Accumulated deficit |
|
|
(36,917 |
) |
|
|
(28,419 |
) |
|
Accumulated other comprehensive loss |
|
|
(588 |
) |
|
|
(533 |
) |
|
Total stockholders' equity |
|
|
2,366 |
|
|
|
10,341 |
|
|
Total liabilities and stockholders' equity |
|
$ |
24,351 |
|
|
$ |
29,034 |
|
The accompanying notes are an integral part of these consolidated financial statements.
F-4
JANONE INC.
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(Dollars in thousands, except per share amounts)
|
|
|
Fiscal Years Ended |
|
|||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Revenues |
|
$ |
33,867 |
|
|
$ |
35,097 |
|
|
Cost of revenues |
|
|
25,040 |
|
|
|
27,311 |
|
|
Gross profit |
|
|
8,827 |
|
|
|
7,786 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
Selling, general and administrative expenses |
|
|
17,823 |
|
|
|
20,217 |
|
|
Operating loss |
|
|
(8,996 |
) |
|
|
(12,431 |
) |
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
Interest expense, net |
|
|
(504 |
) |
|
|
(1,480 |
) |
|
Impairment charges |
|
|
— |
|
|
|
(2,992 |
) |
|
Gain on litigation settlement |
|
|
418 |
|
|
|
694 |
|
|
Gain on settlement of vendor advance payments |
|
|
142 |
|
|
|
— |
|
|
Other income |
|
|
15 |
|
|
|
1,048 |
|
|
Total other income (expense), net |
|
|
71 |
|
|
|
(2,730 |
) |
|
Loss before benefit from income taxes |
|
|
(8,925 |
) |
|
|
(15,161 |
) |
|
Income tax benefit |
|
|
427 |
|
|
|
3,197 |
|
|
Net loss |
|
$ |
(8,498 |
) |
|
$ |
(11,964 |
) |
|
Loss per share: |
|
|
|
|
|
|
|
|
|
Basic loss per share |
|
$ |
(4.59 |
) |
|
$ |
(6.78 |
) |
|
Diluted loss per share |
|
$ |
(4.59 |
) |
|
$ |
(6.78 |
) |
|
Weighted average common shares outstanding: |
|
|
|
|
|
|
|
|
|
Basic |
|
|
1,852,147 |
|
|
|
1,763,670 |
|
|
Diluted |
|
|
1,852,147 |
|
|
|
1,763,670 |
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(8,498 |
) |
|
$ |
(11,964 |
) |
|
Other comprehensive loss, net of tax |
|
|
|
|
|
|
|
|
|
Effect of foreign currency translation adjustments |
|
|
(55 |
) |
|
|
— |
|
|
Total other comprehensive loss, net of tax |
|
|
(55 |
) |
|
|
— |
|
|
Comprehensive loss |
|
$ |
(8,553 |
) |
|
$ |
(11,964 |
) |
The accompanying notes are an integral part of these consolidated financial statements.
F-5
JANONE INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS' EQUITY
(Dollars in thousands)
|
|
|
Series A Preferred |
|
|
Common Stock |
|
|
Additional Paid in |
|
|
Accumulated |
|
|
Accumulated Other Comprehensive |
|
|
|
|
|
|||||||||||||
|
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Deficit |
|
|
Total |
|
||||||||
|
Balance, December 29, 2018 |
|
|
288,588 |
|
|
$ |
— |
|
|
|
1,694,565 |
|
|
$ |
2 |
|
|
$ |
38,660 |
|
|
$ |
(16,518 |
) |
|
$ |
(533 |
) |
|
$ |
21,611 |
|
|
Adoption of ASU 842 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
63 |
|
|
|
— |
|
|
|
63 |
|
|
Share based compensation |
|
|
— |
|
|
|
— |
|
|
|
224,483 |
|
|
|
— |
|
|
|
631 |
|
|
|
— |
|
|
|
— |
|
|
|
631 |
|
|
Shares cancelled |
|
|
(28,859 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(11,964 |
) |
|
|
— |
|
|
|
(11,964 |
) |
|
Balance, December 28, 2019 |
|
|
259,729 |
|
|
$ |
— |
|
|
|
1,919,048 |
|
|
$ |
2 |
|
|
$ |
39,291 |
|
|
$ |
(28,419 |
) |
|
$ |
(533 |
) |
|
$ |
10,341 |
|
|
Effect of foreign currency translation adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(55 |
) |
|
|
(55 |
) |
|
Share based compensation |
|
|
— |
|
|
|
— |
|
|
|
33,191 |
|
|
|
— |
|
|
|
578 |
|
|
|
— |
|
|
|
— |
|
|
|
578 |
|
|
Shares cancelled |
|
|
— |
|
|
|
— |
|
|
|
(122,257 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(8,498 |
) |
|
|
— |
|
|
|
(8,498 |
) |
|
Balance, January 2, 2021 |
|
|
259,729 |
|
|
$ |
— |
|
|
|
1,829,982 |
|
|
$ |
2 |
|
|
$ |
39,869 |
|
|
$ |
(36,917 |
) |
|
$ |
(588 |
) |
|
$ |
2,366 |
|
The accompanying notes are an integral part of these consolidated financial statements.
F-6
JANONE INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(Dollars in thousands)
|
|
|
Fiscal Years Ended |
|
|||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
OPERATING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(8,498 |
) |
|
$ |
(11,964 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
4,122 |
|
|
|
4,076 |
|
|
Amortization of debt issuance costs |
|
|
27 |
|
|
|
307 |
|
|
Stock based compensation expense |
|
|
578 |
|
|
|
631 |
|
|
Gain on settlement of vendor advance payments |
|
|
(142 |
) |
|
|
— |
|
|
Impairment charges |
|
|
— |
|
|
|
2,992 |
|
|
Gain on litigation settlement |
|
|
(418 |
) |
|
|
(694 |
) |
|
Other |
|
|
59 |
|
|
|
131 |
|
|
Changes in assets and liabilities: |
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
|
2,980 |
|
|
|
(765 |
) |
|
Prepaid expenses and other current assets |
|
|
(780 |
) |
|
|
680 |
|
|
Change in deferred rent |
|
|
— |
|
|
|
(48 |
) |
|
Change in deferred compensation |
|
|
— |
|
|
|
(148 |
) |
|
Change in deferred income taxes |
|
|
(270 |
) |
|
|
(3,279 |
) |
|
Income taxes receivable |
|
|
(120 |
) |
|
|
25 |
|
|
Inventories |
|
|
(282 |
) |
|
|
(546 |
) |
|
Right of use assets |
|
|
165 |
|
|
|
(295 |
) |
|
Lease liabilities |
|
|
(73 |
) |
|
|
329 |
|
|
Accounts payable and accrued expenses |
|
|
2,035 |
|
|
|
5,058 |
|
|
Net cash used in operating activities |
|
|
(617 |
) |
|
|
(3,510 |
) |
|
INVESTING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
Purchases of property and equipment |
|
|
(507 |
) |
|
|
(212 |
) |
|
Purchase of intangible asset |
|
|
(327 |
) |
|
|
(288 |
) |
|
Net payments received from ApplianceSmart Holdings LLC note receivable |
|
|
— |
|
|
|
845 |
|
|
Net cash provided by (used in) investing activities |
|
|
(834 |
) |
|
|
345 |
|
|
FINANCING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
Proceeds from related party note |
|
|
— |
|
|
|
2,500 |
|
|
Payment on related party note |
|
|
(1,500 |
) |
|
|
— |
|
|
Proceeds from issuance of short term notes payable |
|
|
3,469 |
|
|
|
471 |
|
|
Payments on short term notes payable |
|
|
(565 |
) |
|
|
(509 |
) |
|
Net cash provided by financing activities |
|
|
1,404 |
|
|
|
2,462 |
|
|
Effect of changes in exchange rate on cash and cash equivalents |
|
|
(55 |
) |
|
|
(11 |
) |
|
DECREASE IN CASH AND CASH EQUIVALENTS |
|
|
(102 |
) |
|
|
(714 |
) |
|
CASH AND CASH EQUIVALENTS, beginning of period |
|
|
481 |
|
|
|
1,195 |
|
|
CASH AND CASH EQUIVALENTS, end of period |
|
$ |
379 |
|
|
$ |
481 |
|
|
|
|
Fiscal Years Ended |
|
|||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Supplemental cash flow disclosures: |
|
|
|
|
|
|
|
|
|
Interest paid |
|
$ |
129 |
|
|
$ |
133 |
|
|
Income taxes paid, net |
|
$ |
30 |
|
|
$ |
263 |
|
The accompanying notes are an integral part of these consolidated financial statements.
F-7
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
The accompanying consolidated financial statements include the accounts of JanOne Inc., a Nevada corporation, and its subsidiaries (collectively the “Company” or “JanOne”). On September 10, 2019, Appliance Recycling Centers of America, Inc. changed its name to JanOne Inc.
The Company has three operating segments – Biotechnology, Recycling, and Technology.
During September 2019, JanOne, through its biotechnology segment, broadened its business perspectives to become a pharmaceutical company focused on finding treatments for conditions that cause severe pain and bringing to market drugs with non-addictive pain-relieving properties.
ARCA Recycling, Inc. (“ARCA Recycling”) provides turnkey recycling services for electric utility energy efficiency programs in the United States. ARCA Canada Inc. (“ARCA Canada”) provides turnkey recycling services for electric utility energy efficiency programs in Canada. Customer Connexx, LLC (“Connexx”) provides call center services for ARCA Recycling and ARCA Canada.
GeoTraq Inc. (“GeoTraq”) is engaged in the development, design and, ultimately, we expect the sale, of cellular transceiver modules, also known as Mobile IoT modules, and associated wireless services.
We report on a 52- or 53-week fiscal year. Our 2020 fiscal year (“2020”) ended on January 2, 2021, and our fiscal year (“2019”) ended on December 28, 2019.
Going concern
The Company currently faces a challenging competitive environment and is focused on improving its overall profitability, which includes managing expenses. We reported a net loss of $8,498 and $11,964 for the fiscal years ended January 2, 2021 and December 28, 2019, respectively. In addition, the Company has as of January 2, 2021 total current assets of $6,941 and total current liabilities $20,597 resulting in a net negative working capital of $13,656.
The Company has available cash balances and funds available under an accounts receivable factoring program with Prestige Capital Finance, LLC (“Prestige Capital”) to provide sufficient liquidity to fund the entity’s operations, the entity’s continued investments in center openings, and remodeling activities for at least the next twelve months. The Company expects to generate cash from operations for the remainder of fiscal year 2021 given its cost cutting measures in response to the revenue reductions resulting from the Coronavirus. However, depending on the U.S.’ continued restrictions related to the coronavirus public health crisis, the Company cannot be certain its efforts will suffice. The agreement with Prestige Capital allows the Company to get advance funding of 80% of an unpaid customer’s invoice amount within 2 days and the balance less a mutually agreed upon fee upon ultimate collection in cash of the invoice. The Company expects that it will be able to utilize the available funds under the accounts receivable factoring agreement to provide liquidity and to pursue acquisitions and other strategic transactions to expand and grow the business to enhance shareholder value. Management also regularly monitors capital market conditions to ensure no other conditions or events exist that may materially affect the Company’s financial conditions and liquidity and the Company may raise additional funds through borrowings or public or private sales of debt or equity securities, if necessary.
On May 1, 2020, the Company entered into a promissory note with Texas Capital Bank, N.A. for $1,872 under the Paycheck Protection Program under the Coronavirus Aid, Relief and Economic Security Act (the “CARES Act”). See Note 14 for a complete discussion about this loan.
Additionally, the Company has $1,500 of availability under its Revolving Credit Facility (as defined in Note 22).
F-8
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
On February 2, 2021, the Offering closed and the Company received gross proceeds of approximately $6,000, before deducting placement agent fees and other offering expenses. The Company is utiizing the net proceeds for general working capital See Note 23 for complete discussion.
Based on the above, management has concluded that as of the filing date on this quarterly report, the Company is not aware and did not identify any other conditions or events that would cause the Company to not be able to continue business as a going concern for the next twelve months.
Coronavirus
In December 2019, the 2019 novel coronavirus (COVID-19) surfaced in Wuhan, China. The World Health Organization declared a global emergency on January 30, 2020, and most countries initiated travel restrictions limiting travel to other countries and lock-downs within their borders. While various vaccines have recently been introduced into the marketplace, the impacts of variant strains of the COVID-19 virus is still unknown. The widespread health crisis has adversely affected the global economy, resulting in an economic downturn that could impact demand for our products. To date, the outbreak had a material adverse impact on our operations. For example, several customers in our appliance recycling and appliance replacement business have previously suspended our ability to pick up and or replace their customers’ appliances resulting in decreased revenues for both recycling and replacement business. The future impact of the outbreak is highly uncertain and cannot be predicted and there is no assurance that the outbreak will not have another material adverse impact on the future results of the Company. The extent of the impact, if any, will depend on future developments, including actions taken to contain the coronavirus. A key task for the Company in 2021 is to begin late-stage clinical development with its pharmaceutical product, JAN101. However, the COVID-19 pandemic significantly impacted clinical trials in 2020, delaying recruitment in most non-COVID-19 clinical trials and even eliminating recruitment in some trials. While clinical sites have largely resumed conducting non-COVID-19 clinical trials, the backlog of subjects may adversely affect our ability to recruit for its trial, leading to longer and more expensive trials. In addition, the unknown effectiveness of the COVID-19 vaccines, particularly concerning variant strains of COVID-19, could lead to clinical sites terminating patient recruitment again during the course of the study.
During April 2020, as a result of the COVID-19 pandemic, the Company entered into an amendment to its contract services agreement with certain customers, whereby those customers agreed to advance the Company $1,168 against the provision of future services. The advanced payment may only be utilized for the costs associated with labor and sustaining ARCA Recycling’s workforce. The advance agreement provides for partial loan forgiveness if certain conditions are met. See Note 14 for a complete discussion of these advances.
|
Note 2: |
Summary of Significant Accounting Policies |
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All significant intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of the consolidated financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumption that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Significant estimates made in connection with the accompanying consolidated financial statements include the estimated reserve for doubtful current and long-term trade and other receivables, the estimated reserve for excess and obsolete inventory, estimated fair value and forfeiture rates for stock-based compensation, fair values in connection with the analysis of other intangibles and long-lived assets for impairment, valuation allowance against deferred tax assets and estimated useful lives for intangible assets and property and equipment.
F-9
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Financial Instruments
Financial instruments consist primarily of cash equivalents, trade and other receivables, notes receivables, and obligations under accounts payable, accrued expenses and notes payable. The carrying amounts of cash equivalents, trade receivables and other receivables, accounts payable, accrued expenses and short-term notes payable approximate fair value because of the short maturity of these instruments. The fair value of the long-term debt is calculated based on interest rates available for debt with terms and maturities similar to the Company’s existing debt arrangements, unless quoted market prices were available (Level 2 inputs). The carrying amounts of long-term debt at January 2, 2021 and December 28, 2019 approximate fair value.
Cash and Cash Equivalents
Cash and cash equivalents consist of highly liquid investments with a maturity of three months or less at the time of purchase. Fair value of cash equivalents approximates carrying value.
Trade Receivables and Allowance for Doubtful Accounts
We carry unsecured trade receivables at the original invoice amount less an estimate made for doubtful accounts based on a monthly review of all outstanding amounts. Management determines the allowance for doubtful accounts by regularly evaluating individual customer receivables and considering a customer’s financial condition, credit history and current economic conditions. We write off trade receivables when we deem them uncollectible. We record recoveries of trade receivables previously written off when we receive them. We consider a trade receivable to be past due if any portion of the receivable balance is outstanding for more than ninety days. We do not charge interest on past due receivables. Our management considers the allowance for doubtful accounts of nil and $29 to be adequate to cover any exposure to loss as of January 2, 2021 and December 28, 2019, respectively.
Inventories
Inventories, consisting primarily of appliances, are stated at the lower of cost, determined on a specific identification basis, or net realizable value. We provide estimated provisions for the obsolescence of our appliance inventories, including adjustment to market, based on various factors, including the age of such inventory and our management’s assessment of the need for such provisions. We look at historical inventory aging reports and margin analyses in determining our provision estimate. A revised cost basis is used once a provision for obsolescence is recorded. The Company does not have a reserve for obsolete inventory at January 2, 2021 and December 28, 2019.
Property and Equipment
Property and Equipment are stated at cost less accumulated depreciation. Expenditures for repairs and maintenance are charged to expense as incurred and additions and improvements that significantly extend the lives of assets are capitalized. Upon sale or other retirement of depreciable property, the cost and accumulated depreciation are removed from the related accounts and any gain or loss is reflected in operations. Depreciation is computed using the straight-line method over the estimated useful lives of the assets. The useful lives of building and improvements are 3 to 30 years, transportation equipment is 3 to 15 years, machinery and equipment are 5 to 10 years, furnishings and fixtures are 3 to 5 years and office and computer equipment are 3 to 5 years.
We periodically review our property and equipment when events or changes in circumstances indicate that their carrying amounts may not be recoverable or their depreciation or amortization periods should be accelerated. We assess recoverability based on several factors, including our intention with respect to maintaining our facilities and projected discounted cash flows from operations. An impairment loss would be recognized for the amount by which the carrying amount of the assets exceeds their fair value, as approximated by the present value of their projected discounted cash flows.
F-10
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Intangible Assets
The Company accounts for intangible assets in accordance with ASC 350, Intangibles—Goodwill and Other. Under ASC 350, intangible assets subject to amortization, shall be reviewed for impairment in accordance with the Impairment or Disposal of Long-Lived Assets in ASC 360, Property, Plant, and Equipment.
Under ASC 360, long-lived assets are tested for recoverability whenever events or changes in circumstances (‘triggering event’) indicate that the carrying amount may not be recoverable. In making this determination, triggering events that were considered included:
|
|
• |
A significant decrease in the market price of a long-lived asset (asset group); |
|
|
• |
A significant adverse change in the extent or manner in which a long-lived asset (asset group) is being used or in its physical condition; |
|
|
• |
A significant adverse change in legal factors or in the business climate that could affect the value of a long-lived asset (asset group), including an adverse action or assessment by a regulator; |
|
|
• |
An accumulation of costs significantly in excess of the amount originally expected for the acquisition or construction of a long-lived asset (asset group); |
|
|
• |
A current-period operating or cash flow loss combined with a history of operating or cash flow losses or a projection or forecast that demonstrates continuing losses associated with the use of a long-lived asset (asset group); and, |
|
|
• |
A current expectation that, more likely than not, a long-lived asset (asset group) will be sold or otherwise disposed of significantly before the end of its previously estimated useful life. The term more likely than not refers to a level of likelihood that is more than 50 percent. |
If a triggering event has occurred, for purposes of recognition and measurement of an impairment loss, a long-lived asset or assets shall be grouped with other assets and liabilities at the lowest level for which identifiable cash flows are largely independent of the cash flows of other assets and liabilities. If after identifying a triggering event it is determined that the asset group’s carrying value may not be recoverable, a recoverability test is performed by forecasting the expected cash flows to be derived from the asset group for the remaining useful life of the asset group’s primary asset compared to its carrying value. The recoverability test relies upon the undiscounted cash flows (excluding interest and taxes) which are derived from the Company’s specific use of those assets (not how a market participant would use those assets); and, are based upon the existing service potential of the current assets (excluding any improvements that would materially enhance the assets). If the expected undiscounted cash flows exceed the carrying value, the assets are considered recoverable.
There was no impairment of intangibles as of January 2, 2021 or December 28, 2019 based on the annual intangible asset impairment test performed as of those dates.
The Company’s intangible assets consist of customer relationship intangibles, trade names, licenses for the use of internet domain names, Universal Resource Locators, or URL’s, software, patent USPTO reference No. 10,182,402, and designs and related manufacturing procedures. Upon acquisition, critical estimates are made in valuing acquired intangible assets, which include but are not limited to: future expected cash flows from customer contracts, customer lists, and estimating cash flows from projects when completed; tradename and market position, as well as assumptions about the period of time that customer relationships will continue; and discount rates. Management's estimates of fair value are based upon assumptions believed to be reasonable, but which are inherently uncertain and unpredictable and, as a result, actual results may differ from the assumptions used in determining the fair values. All intangible assets are capitalized at their original cost and amortized over their estimated useful lives as follows: domain name and marketing – 3 to 20 years; software – 3 to 5 years, technology intangibles – 7 years, customer relationships – 7 to 15 years.
Revenue Recognition
Biotechnology Revenue
We currently are not generating any revenue in our Biotechnology segment.
F-11
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Recycling Revenue
We provide replacement appliances and provide appliance pickup and recycling services for consumers (“end users”) of public utilities, our customers. We receive as part of our de-manufacturing and recycling process revenue from scrap dealers for refrigerant, steel, plastic, glass, copper and other residual items.
We account for revenue in accordance with Accounting Standards Codification 606 Revenue from Contracts with Customers.
Under the revenue standard revenue is recognized as follows:
We determine revenue recognition through the following steps:
|
|
a. |
Identification of the contract, or contracts, with a customer, |
|
|
b. |
Identification of the performance obligations in the contract, |
|
|
c. |
Determination of the transaction price, |
|
|
d. |
Allocation of the transaction price to the performance obligations in the contract, and |
|
|
e. |
Recognition of revenue when, or as, we satisfy a performance obligation. |
As part of its assessment of each contract, the Company evaluates certain factors including the customer’s ability to pay, or credit risk. For each contract, the Company considers the promise to transfer products or services, each of which is distinct, to be the identified performance obligations. In determining the transaction price, the price stated on the contract is typically fixed and represents the net consideration to which the Company expects to be entitled per order, and therefore there is no variable consideration. As the Company’s standard payment terms are less than 90 days, the Company has elected, as a practical expedient, to not assess whether a contract has a significant financing component. The Company allocates the transaction price to each distinct product or service based on its relative standalone selling price. The product or service price as specified on the contract is considered the standalone selling price as it is an observable source that depicts the price as if sold to a similar customer in similar circumstances.
Replacement Product Revenue
We generate revenue by providing replacement appliances. We recognize revenue at the point in time when control over the replacement product is transferred to the end user, when our performance obligations are satisfied, which typically occur upon delivery from our center facility and installation at the end user’s home.
Recycling Services Revenue
We generate revenue by providing pickup and recycling services. We recognize revenue at the point in time when we have picked up a to be recycled appliance and transfer of ownership has occurred, and therefore our performance obligations are satisfied, which typically occur upon pickup from our end user’s home.
Byproduct Revenue
We generate other recycling byproduct revenue (the sale of copper, steel, plastic and other recoverable non-refrigerant byproducts) as part of our de-manufacturing process. We recognize byproduct revenue upon delivery and transfer of control of byproduct to a third-party recycling customer, having a mutually agreed upon price per pound and collection reasonably assured. Transfer of control occurs at the time the customer is in possession of the byproduct material. Revenue recognized is a function of byproduct weight, type and in some cases volume of the byproduct delivered multiplied by the market rate as quoted.
F-12
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Contract Liability
Receivables are recognized in the period we ship the product or provide the service. Payment terms on invoiced amounts are based on contractual terms with each customer. When we receive consideration, or such consideration is unconditionally due, prior to transferring goods or services to the customer under the terms of a sales contract, we record deferred revenue, which represents a contract liability. We recognize a contract liability as net sales once control of goods and/or services have been transferred to the customer and all revenue recognition criteria have been met and any constraints have been resolved. We defer the product costs until recognition of the related revenue occurs.
Assets Recognized from Costs to Obtain a Contract with a Customer
We recognize an asset for the incremental costs of obtaining a contract with a customer if it expects the benefit of those costs to be longer than one year. We have concluded that no material costs have been incurred to obtain and fulfill our FASB Accounting Standards Codification, or ASC 606 contracts, meet the capitalization criteria, and as such, there are no material costs deferred and recognized as assets on the consolidated balance sheet at January 2, 2021 or December 28, 2019.
Other:
|
|
a. |
Taxes collected from customers and remitted to government authorities and that are related to sales of our products are excluded from revenues. |
|
|
b. |
Sales commissions are expensed when incurred because the amortization period would have been one year or less. These costs are recorded in Selling, General and Administrative expense. |
|
|
c. |
We do not disclose the value of unsatisfied performance obligations for (i) contracts with original expected lengths of one year or less or (ii) contracts for which we recognize revenue at the amount to which we have the right to invoice for the services performed. |
Revenue recognized for Company contracts - $31,518 and $32,136 for the years ended January 2, 2021 and December 28, 2019, respectively. Byproduct revenue is non-contract revenue and amounts for Byproduct revenue have been excluded from Revenue recognized for Company contracts for all periods presented.
Technology Revenue
We currently are not generating any revenue from our Technology segment.
Shipping and Handling
The Company classifies shipping and handling charged to customers as revenues and classifies costs relating to shipping and handling as cost of revenues.
Advertising Expense
Advertising expense is charged to operations as incurred. Advertising expense was $379 and $827 for the years ended January 2, 2021 and December 28, 2019, respectively.
F-13
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Fair Value Measurements
ASC Topic 820, “Fair Value Measurements and Disclosures,” requires disclosure of the fair value of financial instruments held by the Company. ASC Topic 825, “Financial Instruments,” defines fair value, and establishes a three-level valuation hierarchy for disclosures of fair value measurement that enhances disclosure requirements for fair value measures. The three levels of valuation hierarchy are defined as follows: Level 1 - inputs to the valuation methodology are quoted prices for identical assets or liabilities in active markets. Level 2 – to the valuation methodology include quoted prices for similar assets and liabilities in active markets, and inputs that are observable for the asset or liability, either directly or indirectly, for substantially the full term of the financial instrument. Level 3 – inputs to the valuation methodology are unobservable and significant to the fair value measurement.
Income Taxes
The Company accounts for income taxes using the asset and liability method. The asset and liability method requires recognition of deferred tax assets and liabilities for expected future tax consequences of temporary differences that currently exist between tax bases and financial reporting bases of the Company's assets and liabilities. Deferred income tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which these temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance is provided on deferred taxes if it is determined that it is more likely than not that the asset will not be realized. The Company recognizes penalties and interest accrued related to income tax liabilities in the provision for income taxes in its Consolidated Statements of Income.
Significant management judgment is required to determine the amount of benefit to be recognized in relation to an uncertain tax position. The Company uses a two-step process to evaluate tax positions. The first step requires an entity to determine whether it is more likely than not (greater than 50% chance) that the tax position will be sustained. The second step requires an entity to recognize in the financial statements the benefit of a tax position that meets the more-likely-than-not recognition criterion. The amounts ultimately paid upon resolution of issues raised by taxing authorities may differ materially from the amounts accrued and may materially impact the financial statements of the Company in future periods.
Lease Accounting
We account for leases in accordance with ASC 842 - Leases This accounting standard requires all lessees to record the impact of leasing contracts on the balance sheet as a right to use asset and corresponding liability. This is measured by taking the present value of the remaining lease payments over the lease term and recording a right to use asset (“ROU”) and corresponding lease obligation for lease payments. Rent expense is realized on a straight-line basis and the lease obligation is amortized based on the effective interest method. The amounts recognized reflect the present value of remaining lease payments for all leases that have a lease term greater than 12 months. The discount rate used is an estimate of the Company’s incremental borrowing rate based on information available at lease commencement.
In considering the lease asset value, the Company considers fixed or variable payment terms, prepayments and options to extend, terminate or purchase. Renewal, termination or purchase options affect the lease term used for determining lease asset value only if the option is reasonably certain to be exercised. The Company uses an estimate of its incremental borrowing rate based on information available at lease commencement in determining present value of lease payments.
We lease warehouse facilities and office space. These assets and properties are generally leased under noncancelable agreements that expire at various dates through 2025 with various renewal options for additional periods. The agreements, which have and continue to be classified as operating leases, generally provide for base rent and require us to pay all insurance, taxes and other maintenance costs. The Company’s operating leases are exclusively for building space in the different cities we have operations. The lease terms typically last from 2-5 years with some being longer or shorter depending on needs of the business and the lease partners. The Company has also engaged in month-to-month leases for parking spaces that the Company has elected to expense as incurred. Our lease
F-14
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
agreements do not include variable lease payments. Our lessors do offer options to extend lease terms as leases expire and management evaluates against current rental markets and other strategic factors in making the decision to renew. When leases are within 6 months of being renewed, management will estimate probabilities of renewing for an additional term based on market and strategic factors and if the probability is more likely than not that the lease will be renewed, the financials will assume the lease is renewed under the lease renewal option.
The operating leases we have do not contain residual value guarantees and do not contain restrictive covenants. The Company currently has one sublease in Ontario, Canada.
Leases accounted under ASC 842 were determined based on analysis of the lease contracts using lease payments and timing as documented in the contract. Non lease contracts were also evaluated to understand if the contract terms provided for an asset that we controlled and provided us with substantially all the economic benefits. We did not observe any contracts with embedded leases. Lease contracts were reviewed, and distinctions made between non lease and lease payments. Only payments related to the lease of the asset were included in lease payment calculations. Management uses an estimation of its incremental borrowing rate at lease commencement over similar terms as the lease contracts in determining the present value of its lease obligations.
The Company from time to time grants stock options to employees, non-employees and Company executives and directors. Such awards are valued based on the grant date fair-value of the instruments, net of estimated forfeitures. The value of each award is amortized on a straight-line basis over the vesting period.
Foreign Currency
The financial statements of the Company’s non-U.S. subsidiary are translated into U.S. dollars in accordance with ASC 830, Foreign Currency Matters. Under ASC 830, if the assets and liabilities of the Company are recorded in certain non-U.S. functional currencies other than the U.S. dollar, they are translated at rates of exchange at year end. Revenue and expense items are translated at the average monthly exchange rates. The resulting translation adjustments are recorded directly into accumulated other comprehensive income (loss).
Earnings Per Share
Earnings per share is calculated in accordance with ASC 260, “Earnings Per Share”. Under ASC 260 basic earnings per share is computed using the weighted average number of common shares outstanding during the period except that it does not include unvested restricted stock subject to cancellation. Diluted earnings per share is computed using the weighted average number of common shares and, if dilutive, potential common shares outstanding during the period. Potential common shares consist of the incremental common shares issuable upon the exercise of warrants, options, restricted shares and convertible preferred stock. The dilutive effect of outstanding restricted shares, options and warrants is reflected in diluted earnings per share by application of the treasury stock method. Convertible preferred stock is reflected on an if-converted basis.
Segment Reporting
ASC Topic 280, “Segment Reporting,” requires use of the “management approach” model for segment reporting. The management approach model is based on the way a Company’s management organizes segments within the Company for making operating decisions and assessing performance. The Company determined it has three reportable segments (See Note 21).
F-15
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Concentration of Credit Risk
The Company maintains cash balances at several banks in several states including, California, Minnesota and Nevada. Accounts are insured by the Federal Deposit Insurance Corporation up to $250 per institution. At times, balances may exceed federally insured limits.
Recently Issued Accounting Pronouncements
In June 2016, the Financial Accounting Standards Board issued ASU No. 2016-13, Measurement of Credit Losses on Financial Instruments, which introduces a new approach to estimate credit losses on certain types of financial instruments based on expected losses instead of incurred losses. It also modifies the impairment model for available-for-sale debt securities and provides a simplified accounting model for purchased financial assets with credit deterioration since their origination. ASU No. 2016-13 is effective for smaller reporting companies for fiscal years beginning after December 15, 2022 and interim periods within those fiscal years. Early adoption is permitted. We are currently assessing the impact of adopting this new accounting standard on our Consolidated Financial Statements and related disclosures.
In December 2019, the FASB issued ASU No. 2019-12 - Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes (“ASU 2019-12”). ASU 2019-12 is part of the FASB’s overall simplification initiative and seeks to simplify the accounting for income taxes by updating certain guidance and removing certain exceptions. The updated guidance is effective for fiscal years beginning after December 15, 2020 and interim periods within those fiscal years. Early adoption is permitted. The Company is currently assessing the impact of adopting this new accounting standard on its consolidated financial statements and related disclosures.
In March 2020, the FASB issued ASU No. 2020-04 - Reference Rate Reform (Topic 848), codified as ASC 848 (“ASC 848”). The purpose of ASC 848 is to provide optional guidance to ease the potential effects on financial reporting of the market-wide migration away from Interbank Offered Rates to alternative reference rates. ASC 848 applies only to contracts, hedging relationships, and other transactions that reference a reference rate expected to be discontinued because of reference rate reform. The guidance may be applied upon issuance of ASC 848 through December 31, 2022. The Company is currently assessing the impact of adopting this new accounting standard on its consolidated financial statements and related disclosures.
Note 3: Trade and other receivables
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Trade receivables, net |
|
$ |
4,174 |
|
|
$ |
7,226 |
|
|
Factored accounts receivable |
|
|
(891 |
) |
|
|
(2,165 |
) |
|
Prestige Capital reserve receivable |
|
|
162 |
|
|
|
415 |
|
|
Due from Recleim |
|
|
— |
|
|
|
913 |
|
|
Other receivables |
|
|
155 |
|
|
|
189 |
|
|
Trade and other receivables, net |
|
$ |
3,600 |
|
|
$ |
6,578 |
|
|
|
|
|
|
|
|
|
|
|
|
Trade accounts receivable |
|
$ |
2,698 |
|
|
$ |
5,928 |
|
|
Un-billed trade receivables |
|
|
1,476 |
|
|
|
1,327 |
|
|
Accounts receivable reserve |
|
|
— |
|
|
|
(29 |
) |
|
Total trade receivables, net |
|
$ |
4,174 |
|
|
$ |
7,226 |
|
F-16
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
|
Note 4: |
Note receivable |
On December 30, 2017, we signed an agreement to dispose of our retail appliance segment. ApplianceSmart Holdings LLC (the “Purchaser”), a wholly owned subsidiary of Live Ventures Incorporated, entered into a Stock Purchase Agreement (the “Agreement”) with the Company and ApplianceSmart, then a subsidiary of the Company. ApplianceSmart is a retail chain specializing in new and out-of-the-box appliances. Pursuant to the Agreement, the Purchaser purchased from the Company all the issued and outstanding shares of capital stock (the “Stock”) of ApplianceSmart in exchange for $6,500 (the “Purchase Price”). The Purchase Price per the Agreement was due and payable on or before March 31, 2018.
Between March 31, 2018 and April 24, 2018, the Purchaser and the Company negotiated in good faith the method of payment of the remaining outstanding balance of the Purchase Price. On April 25, 2018, the Purchaser delivered to the Company a promissory note (the “ApplianceSmart Note”) in the original principal amount of $3,919 (the “Original Principal Amount”), as such amount may be adjusted per the terms of the ApplianceSmart Note. The ApplianceSmart Note is effective as of April 1, 2018 and matures on April 1, 2021 (the “Maturity Date”). The ApplianceSmart Note bears interest at 5% per annum with interest and principal payable at the Maturity Date. ApplianceSmart provided the Company a guaranty of repayment of the ApplianceSmart Note. The remaining $2,581 of the Purchase Price was paid in cash by the Purchaser to the Company. The Purchaser may reborrow funds, and pay interest on such re-borrowings, from the Company up to the Original Principal Amount. Subsequent to December 30, 2017, ApplianceSmart assumed $1,901 in liabilities from the Company. For the 52 weeks ended December 29, 2018, the original balance owed to the Company of $6,500, increased with new borrowings of $1,819 and decreased with repayments of $2,581 and debt assumed of $1,901 represents a net amount due from the Purchaser, now in the form of a note receivable.
On December 26, 2018, the ApplianceSmart Note was amended and restated to grant the Company a security interest in the assets of the Purchaser, ApplianceSmart, and ApplianceSmart Contracting Inc. in exchange for modifying the repayments terms to provide for the payment in full of all accrued interest and principal on the Maturity Date of the ApplianceSmart Note.
On March 15, 2019, the Company entered into agreements with third parties pursuant to which it agreed to subordinate the payment of indebtedness under the ApplianceSmart Note and the Company’s security interest in the assets of ApplianceSmart in exchange for a prepayment of up to $1,200. Additionally, the Company advanced ApplianceSmart $355 during fiscal 2019 under the ApplianceSmart Note.
On December 9, 2019, ApplianceSmart filed a voluntary petition in the United States Bankruptcy Court for the Southern District of New York seeking relief under Chapter 11 of Title 11 of the United States Code. As a result, the Company has recorded an impairment charge of $2,992 for the amount owed by ApplianceSmart to the Company as of December 28, 2019. The outstanding balance of the ApplianceSmart Note at January 2, 2021 and December 28, 2019 was $2,992 and $2,992, respectively, exclusive of the impairment charge.
|
Note 5: |
Inventory |
Inventories of continuing operations, consisting principally of appliances, are stated at the lower of cost, determined on a specific identification basis, or net realizable value and consist of the following as of January 2, 2021 and December 28, 2019:
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Appliances held for resale |
|
$ |
1,430 |
|
|
$ |
1,148 |
|
|
Raw material - chips |
|
|
200 |
|
|
|
200 |
|
|
Total inventory |
|
$ |
1,630 |
|
|
$ |
1,348 |
|
We provide estimated provisions for the obsolescence of our appliance inventories, as necessary, including adjustments to net realizable value, based on various factors, including the age of such inventory and our management’s assessment of the need for such provisions. We look at historical inventory aging reports and margin analyses in determining our provision estimate. A revised cost basis is used once a provision for obsolescence is recorded.
F-17
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
|
Note 6: |
Prepaids and other current assets |
Prepaids and other current assets as of January 2, 2021 and December 28, 2019 consist of the following:
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Prepaid insurance |
|
$ |
371 |
|
|
$ |
282 |
|
|
Prepaid rent |
|
|
95 |
|
|
|
— |
|
|
Prepaid purchase orders |
|
|
366 |
|
|
|
— |
|
|
Prepaid other |
|
|
304 |
|
|
|
74 |
|
|
|
|
$ |
1,136 |
|
|
$ |
356 |
|
|
Note 7: |
Property and equipment |
Property and equipment of continuing operations as of January 2, 2021 and December 28, 2019 consist of the following:
|
|
|
Useful Life (Years) |
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Buildings and improvements |
|
3-30 |
|
$ |
75 |
|
|
$ |
69 |
|
|
Equipment |
|
3-15 |
|
|
2,528 |
|
|
|
2,314 |
|
|
Projects under construction |
|
|
|
|
387 |
|
|
|
120 |
|
|
Property and equipment |
|
|
|
|
2,990 |
|
|
|
2,503 |
|
|
Less accumulated depreciation |
|
|
|
|
(2,258 |
) |
|
|
(2,179 |
) |
|
Property and equipment, net |
|
|
|
$ |
732 |
|
|
$ |
324 |
|
Depreciation expense was $79 and $99 for fiscal years 2020 and 2019, respectively.
|
Note 8: |
Intangible assets |
Intangible assets as of January 2, 2021 and December 28, 2019 consist of the following:
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Intangible assets GeoTraq |
|
$ |
26,096 |
|
|
$ |
26,096 |
|
|
Patents and domains |
|
|
23 |
|
|
|
23 |
|
|
Computer software |
|
|
4,494 |
|
|
|
4,167 |
|
|
|
|
|
30,613 |
|
|
|
30,286 |
|
|
Less accumulated amortization |
|
|
(16,624 |
) |
|
|
(12,581 |
) |
|
|
|
$ |
13,989 |
|
|
$ |
17,705 |
|
The useful life and amortization period of the GeoTraq intangible acquired is seven years. Intangible amortization expense for continuing operations was $4,043 and $3,977 for fiscal years 2020 and 2019, respectively.
|
Note 9: |
Deposits and other assets |
Deposits and other assets as of January 2, 2021 and December 28, 2019 consist of the following:
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Deposits |
|
$ |
169 |
|
|
$ |
195 |
|
|
Other |
|
|
62 |
|
|
|
77 |
|
|
|
|
$ |
231 |
|
|
$ |
272 |
|
Deposits are primarily refundable security deposits with landlords the Company leases property from.
F-18
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Note 10: Leases
We account for leases in accordance with ASC 842. The amount recorded is the present value of all remaining lease payments for leases with terms greater than 12 months. The right of use asset is offset by a corresponding liability. The discount rate is based on an estimate of our incremental borrowing rate for terms similar to our lease terms at the time of lease commencement. The asset will be amortized over remaining lease terms. See Lease Accounting in Note 2.
Total present value of future lease payments as of January 2, 2021:
|
2021 |
|
$ |
1,347 |
|
|
2022 |
|
|
733 |
|
|
2023 |
|
|
436 |
|
|
2024 |
|
|
305 |
|
|
2025 |
|
|
47 |
|
|
Total |
|
|
2,868 |
|
|
Less interest |
|
|
(283 |
) |
|
Present value of payments |
|
$ |
2,585 |
|
During the years ended January 2, 2021 and December 28, 2019, $1,250 and $1,284, respectively, was included in operating cash flow for amounts paid for operating leases.
Additionally, we obtained right-of-use assets in exchange for lease liabilities of approximately $1,367 upon commencement of operating leases during the year ended January 2, 2021. Additionally, we exercised an early termination clause in one our leases which reduces our right of use assets by $234.
The weighted average lease term for operating leases is 2.7 years and the weighted average discount rate is 8%.
Note 11: Accrued liabilities
Accrued liabilities of continuing operations as of January 2, 2021 and December 28, 2019 consist of the following:
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Compensation and benefits |
|
$ |
604 |
|
|
$ |
809 |
|
|
Contract liability |
|
|
292 |
|
|
|
515 |
|
|
Accrued incentive and rebate checks |
|
|
1,220 |
|
|
|
988 |
|
|
Accrued rent |
|
|
— |
|
|
|
228 |
|
|
Accrued transportation costs* |
|
|
662 |
|
|
|
— |
|
|
Accrued guarantees |
|
|
767 |
|
|
|
767 |
|
|
Accrued purchase orders |
|
|
177 |
|
|
|
— |
|
|
Accrued taxes |
|
|
299 |
|
|
|
297 |
|
|
Other |
|
|
867 |
|
|
|
334 |
|
|
|
|
$ |
4,888 |
|
|
$ |
3,938 |
|
*Accrued transportation costs are related to delayed billing from certain vendors.
F-19
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Contract liabilities rollforward
The following table summarizes the contract liability activity for the year ended January 2, 2021:
|
Beginning balance, December 28, 2019 |
|
$ |
515 |
|
|
Accrued |
|
|
649 |
|
|
Settled |
|
|
(872 |
) |
|
Ending balance, January 2, 2021 |
|
$ |
292 |
|
Note 12: Accrued liability – California sales tax
We operate in fourteen states in the U.S. and in various provinces in Canada. From time to time, we are subject to sales and use tax audits that could result in additional taxes, penalties and interest owed to various taxing authorities.
The California Department of Tax and Fee Administration (formerly known as the California Board of Equalization) (“CDTFA”) conducted a sales and use tax examination covering ARCA Recycling’s California operations for years 2011, 2012 and 2013. The Company believed it was exempt from collecting sales taxes under service agreements with utility customers that included appliance replacement programs. During the fourth quarter of 2014, the Company received communication from the CDTFA indicating they were not in agreement with the Company’s interpretation of the law. As a result, the Company applied for and, as of February 9, 2015, received approval to participate in the CDTFA’s Managed Audit Program. The period covered under this program included years 2011, 2012, 2013 and extended through the nine-month period ended September 30, 2014.
On April 13, 2017 the Company received the formal CDTFA assessment for sales tax for tax years 2011, 2012 and 2013 in the amount of $4,132 plus applicable interest of approximately $500 related to the appliance replacement programs that the Company administered on behalf of its customers on which it did not assess, collect or remit sales tax. The Company has appealed this assessment to the CDTFA Appeals Bureau. The appeal remains in process. Interest continues to accrue until the matter is settled.
As of January 2, 2021 and December 28, 2019, our accrued liability for California sales tax was $5,769 and $5,438, respectively.
|
Note 13: |
Income taxes |
For fiscal year 2020 and 2019, we recorded an income tax benefit of $427 and $3,197, respectively, which consisted of the following:
|
|
|
Fiscal Years Ended |
|
|||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Current tax expense: |
|
|
|
|
|
|
|
|
|
State |
|
$ |
(46 |
) |
|
$ |
(80 |
) |
|
Federal |
|
|
203 |
|
|
|
— |
|
|
Current tax expense |
|
$ |
157 |
|
|
$ |
(80 |
) |
|
Deferred tax benefit - domestic |
|
|
270 |
|
|
|
3,277 |
|
|
Benefit of income taxes |
|
$ |
427 |
|
|
$ |
3,197 |
|
F-20
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
A reconciliation of our benefit of income taxes with the federal statutory tax rate for fiscal years 2020 and 2019 is shown below:
|
|
|
Fiscal Years Ended |
|
|||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
U.S statutory rate |
|
|
21.0 |
% |
|
|
21.0 |
% |
|
State tax rate |
|
|
7.5 |
% |
|
|
1.8 |
% |
|
Foreign rate differential |
|
|
0.4 |
% |
|
|
0.2 |
% |
|
Permanent differences |
|
|
-0.1 |
% |
|
|
-0.1 |
% |
|
Change in valuation allowance |
|
|
-24.5 |
% |
|
|
-0.7 |
% |
|
Other |
|
|
0.6 |
% |
|
|
-0.6 |
% |
|
|
|
|
4.9 |
% |
|
|
21.6 |
% |
Loss before benefit of income taxes was derived from the following sources for fiscal years 2020 and 2019 as shown below:
|
|
|
Fiscal Years Ended |
|
|||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
United States |
|
$ |
(8,270 |
) |
|
$ |
(14,497 |
) |
|
Canada |
|
|
(655 |
) |
|
|
(664 |
) |
|
|
|
$ |
(8,925 |
) |
|
$ |
(15,161 |
) |
The components of net deferred tax assets (liabilities) as of January 2, 2021 and December 28, 2019, are as follows:
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Deferred tax assets (liabilities): |
|
|
|
|
|
|
|
|
|
Allowance for bad debts |
|
$ |
823 |
|
|
$ |
802 |
|
|
Accrued expenses |
|
|
1,769 |
|
|
|
1,623 |
|
|
Accrued compensation |
|
|
76 |
|
|
|
62 |
|
|
Prepaid expenses |
|
|
(304 |
) |
|
|
(93 |
) |
|
Net operating loss |
|
|
3,477 |
|
|
|
2,045 |
|
|
Lease liability |
|
|
35 |
|
|
|
504 |
|
|
Tax credits |
|
|
290 |
|
|
|
256 |
|
|
Share-based compensation |
|
|
176 |
|
|
|
125 |
|
|
Intangibles |
|
|
(3,717 |
) |
|
|
(4,585 |
) |
|
Property and equipment |
|
|
(214 |
) |
|
|
(652 |
) |
|
Unrealized losses (gains) |
|
|
140 |
|
|
|
141 |
|
|
Section 163(j) interest |
|
|
387 |
|
|
|
288 |
|
|
|
|
|
2,938 |
|
|
|
516 |
|
|
Less: valuation allowance |
|
|
(2,938 |
) |
|
|
(786 |
) |
|
Net deferred tax assets (liabilities) |
|
$ |
— |
|
|
$ |
(270 |
) |
As of January 2, 2021, the Company has net operating loss carryforwards of approximately $10,221 for federal income tax purposes, which will be available to offset future taxable income. Due to recent tax legislation, these net operating losses are eligible for indefinite carryforward, limited by certain taxable income limitations. The Company evaluates all available evidence to determine if a valuation allowance is needed to reduce its deferred tax assets. Management has concluded that it is more likely than not that its existing tax benefits in the U.S. and Canada will not be realized. Accordingly, the Company has recorded a valuation allowance $2,938 at January 2, 2021 to reduce its deferred tax assets.
F-21
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
The Company annually conducts an analysis of its uncertain tax positions and has concluded that it has no uncertain tax positions as of January 2, 2021. The Company’s policy is to record uncertain tax positions as a component of income tax expense.
The Company files U.S. and state income tax returns in jurisdictions with differing statutes of limitations. The 2017 through 2020 tax years remain subject to selection for examination as of January 2, 2021. None of the Company’s income tax returns are currently under audit.
|
Note 14: |
Short term debt |
Short term debt and other financing obligations as of January 2, 2021 and December 28, 2019, consist of the following:
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
AFCO Finance |
|
$ |
144 |
|
|
$ |
155 |
|
|
GE 8% loan agreement (Note 15) |
|
|
— |
|
|
|
125 |
|
|
Payroll protection program |
|
|
1,872 |
|
|
|
— |
|
|
Vendor advance payments |
|
|
1,026 |
|
|
|
— |
|
|
Total short term debt |
|
$ |
3,042 |
|
|
$ |
280 |
|
AFCO Finance
Annually, we enter into a financing agreement with AFCO Credit Corporation (“AFCO”) purchased through Marsh Insurance to fund the annual premiums on insurance policies due June 1 of each year. These policies are related to workers’ compensation and various liability policies including, but not limited to, General, Auto, Umbrella, Property, and Directors’ and Officers’ insurance. The total amount of the premiums financed during June 2020 was $429 with an interest rate of 3.3%. An initial down payment of $143 was due before July 1, 2020 with additional monthly payments of $48 made beginning July 1, 2020 and ending June 1, 2021.
The outstanding principal due AFCO at January 2, 2021 and December 28, 2019 was $144 and $155, respectively.
Payroll Protection Program
On May 1, 2020, the Company entered into a promissory note (the “Promissory Note”) with Texas Capital Bank, N.A. that provides for a loan in the amount of $1,872 (the “PPP Loan”) pursuant to the Paycheck Protection Program under the Coronavirus Aid, Relief and Economic Security Act (the “CARES Act”). The PPP Loan matures on April 27, 2022 and bears interest at a rate of 1.0% per annum. Monthly amortized principal and interest payments are deferred for six months after the date of disbursement. The Promissory Note contains events of default and other provisions customary for a loan of this type. The Paycheck Protection Program provides that the use of the PPP Loan amount shall be limited to certain qualifying expenses and may be partially or wholly forgiven in accordance with the requirements set forth in the CARES Act. The Company has applied for forgiveness of a portion of the loan in accordance with the terms of the CARES Act to the extent applicable.
Customer Advance Payments
As of the period ending June 27, 2020, the Company received advance payments authorized by the California Public Utilities Commission and processed through two California utilities for the purposes of sustaining the workforce during the COVID-19 pandemic shutdown. The use of these funds was limited to labor and labor benefits for impacted employees. Portions of these advances are forgivable if certain conditions are met the specifics which have not been finalized. Advance payments that are not forgiven will need to be paid back in full by December 31, 2021. Total funding received under this program as of September 26, 2020 amounted to $1,168. As of January 2, 2021, $142 was forgiven, leaving a balance of $1,026.
F-22
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
|
Note 15: |
Commitments and Contingencies |
Litigation
On December 29, 2016, the Company served a Minnesota state court complaint for breach of contract on Skybridge Americas, Inc. (“SA”), the Company’s primary call center vendor throughout 2015 and most of 2016. The Company seeks damages in the millions of dollars as a result of alleged overcharging by SA and lost client contracts. On January 25, 2017, SA served a counterclaim for unpaid invoices in the amount of approximately $460 plus interest and attorneys’ fees. On March 29, 2017, the Hennepin County district court (the “District Court”) dismissed the Company’s breach of contract claim based on SA’s overuse of its Canadian call center but permitted the Company’s remaining claims to proceed. Following motion practice, on January 8, 2018 the District Court entered judgment in SA’s favor, which was amended as of February 28, 2018, for a total amount of $614, including interest and attorneys’ fees. On March 4, 2019, the Minnesota Court of Appeals (the “Court of Appeals”) ruled and (i) reversed the District Court’s judgment in favor of Skybridge on the call center location claim and remanded the issue back to the District Court for further proceedings, (ii) reversed the District Court’s judgment in favor of Skybridge on the net payment issue and remanded the issue to the District Court for further proceedings, and (iii) affirmed the District Court’s judgment in Skybridge’s favor against the Company’s claim that Skybridge breached the contract when it failed to meet the service level agreements. As a result of the decision by the Court of Appeals, the District Court’s award of interest and attorneys’ fees, etc. was reversed. The Company and SA held a mediation session in July 2020. Trial was held in August 2020 and on February 1, 2021, the District Court awarded damages against the Company in the amount of $715, plus interest, fees, and costs. The Company filed a motion for a new trial and is waiting for the District Court to rule.
On November 15, 2016, the Company served an arbitration demand on Haier US Appliance Solutions, Inc., dba GE Appliances (“GEA”), alleging breach of contract and interference with prospective business advantage. The Company seeks over $2,000 in damages. On April 18, 2017, GEA served a counterclaim for approximately $337 in alleged obligations under the parties’ recycling agreement. Simultaneously with serving its counterclaim in the arbitration, which is venued in Chicago, GEA filed a complaint in the United States District Court for the Western District of Kentucky seeking damages of approximately $530 plus interest and attorneys’ fees allegedly owed under a previous agreement between the parties. On December 12, 2017, the court stayed GEA’s complaint in favor of the arbitration. Under the terms of the Company’s transaction with Recleim LLC (“Recleim”), Recleim is obligated to pay GEA on the Company’s behalf the amounts claimed by GEA in the arbitration and in the lawsuit pending in Kentucky. Those amounts have been paid into escrow pending the outcome of the arbitration. Arbitration proceedings were held in October and November 2019. On March 5, 2020, the arbitrator ruled in part in favor of the Company and in part in favor of GEA, and, as a result, GEA was awarded approximately $125 in damages.
AMTIM Capital, Inc. (“AMTIM”) acts as the Company’s representative to market our recycling services in Canada under an arrangement that pays AMTIM for revenues generated by recycling services in Canada as set forth in the agreement between the parties. A dispute has arisen between AMTIM and the Company with respect to the calculation of amounts due to AMTIM pursuant to the agreement. In a lawsuit filed in the province of Ontario, AMTIM claims a discrepancy in the calculation of fees due to AMTIM by the Company of approximately $2,000. Although the outcome of this claim is uncertain, the Company believes that no further amounts are due under the terms of the agreement and that we will continue to defend our position relative to this lawsuit. Trial is currently scheduled for April 2021.
On October 4, 2018, the Company initiated litigation against a former professional services provider (“PSP”), in Illinois state court, as well as a private arbitration proceeding that was scheduled to be held in Minneapolis, Minnesota, arising from PSP’s rendering of certain professional services to the Company during the period from 2011 through 2014. PSP filed a counterclaim in the arbitration seeking an award of its legal fees and costs arising from that proceeding. The parties subsequently agreed to consolidate their respective claims into the arbitration. The Company’s arbitration demand, as amended, sought an award of more than $50 and other relief. On March 23, 2020, the parties entered into a settlement agreement, whereby, without any admission of liability, they exchanged mutual releases, agreed to dismiss their respective claims with prejudice, and PSP agreed to pay $800 to the Company to, among other things, assist it with certain of its costs and obligations that related to various issues underlying the arbitration proceeding.
F-23
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Other Commitments
As previously disclosed and as discussed in Note 4: Note receivable, on December 30, 2017, the Company disposed of its retail appliance segment and sold ApplianceSmart to the Purchaser. In connection with that sale, as of January 2, 2021 the Company has an aggregate amount of future real property lease payments of $767, which represents amounts guaranteed or which may be owed under certain lease agreements to third party landlords in which the Company either remains the counterparty, is a guarantor, or has agreed to remain contractually liable under the lease (“ApplianceSmart Leases”).
The Company evaluated the fair value of its potential obligation under the guidance of ASC 450: Contingencies and ASC 460: Guarantees. As a result, the Company accrued the amount of liability associated with these future guaranteed lease payments. The fair value was calculated based on the amounts reported as part of the bankruptcy proceedings as ApplianceSmart terminated the leases prior to the lease termination date. The fair value was calculated based on the undiscounted lease payments, a discount rate equivalent to current interest rates associated with the leased real estate and a remote probability weighting of 1%.
The ApplianceSmart Leases either have the Company as the contract tenant only, or in the contract reflects a joint tenancy with ApplianceSmart. ApplianceSmart is the occupant of the ApplianceSmart Leases. The Company does not have the right to use the ApplianceSmart lease assets nor is the Company the primary obligor of the lease payments, hence capitalization under ASC 840 is not required. The ApplianceSmart Leases have historically been used by ApplianceSmart for their operations and the consideration has and is being paid by ApplianceSmart historically and in the future.
Any potential amounts paid out for the Company obligations and or guarantees under ApplianceSmart Leases would be recoverable to the extent there are assets available from ApplianceSmart. ApplianceSmart Leases are related party transactions. The Company divested itself of the ApplianceSmart Leases and leaseholds with the sale to Purchaser on December 30, 2017.
The Company is party from time to time to other ordinary course disputes that we do not believe to be material to our financial condition as of January 2, 2021.
|
Note 16: |
Series A-1 Preferred Stock |
On August 18, 2017, the Company acquired GeoTraq by way of merger, resulting in GeoTraq becoming a wholly-owned subsidiary of the Company. In connection with this transaction, the Company tendered to the owners of GeoTraq $200 cash, issued to them an aggregate of 288,588 shares of the Company’s Series A Convertible Preferred Stock (the “Series A Preferred Stock”), and entered into one-year unsecured promissory notes in the aggregate principal amount of $800.
To accomplish the designation and issuance of the Series A Preferred Stock, we filed a Certificate of Designation with the Secretary of State of the State of Minnesota. On November 9, 2017, we filed a Certificate of Correction with the Minnesota Secretary of State. In connection with the Reincorporation, we filed Articles of Incorporation with the Secretary of State of the State of Nevada on March 12, 2018, and a Certificate of Correction with the Secretary of State of the State of Nevada on August 7, 2018 (collectively, the “Nevada Articles of Incorporation”). On June 21, 2019, we filed a Certificate of Designation of Powers, Preferences, and Rights of Series A-1 Convertible Preferred Stock (the “Series A-1 Preferred Stock”) with the Nevada Secretary of State. On October 1, 2020, we filed an Amended and Restated Certificate of Designation for the Preferences, Rights, and Limitations of the Series A-1 Convertible Preferred Stock (the “Amended and Restated Series A-1 Certificate of Designation”) with the Nevada Secretary of State. The following summary of the Nevada Articles of Incorporation and Amended and Restated Series A-1 Certificate of Designation does not purport to be complete and is qualified in its entirety by reference to the provisions of applicable law and to the Nevada Articles of Incorporation and the Amended and Restated Series A-1 Certificate of Designation, which are filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K filed with the SEC on March 13, 2018, as Exhibit 3.1. to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2018, and Exhibit 3.8(a) to the Company’s Current Report on Form 8-K filed with the SEC on October 2, 2020.
F-24
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
The Series A-1 Convertible Preferred Stock was designated pursuant to guidance received from Nasdaq and has virtually all of the same rights, characteristics, and attributes as the Company’s Series A Convertible Preferred Stock, except as required by the Listing Qualifications staff of The Nasdaq Stock Market LLC (i.e., Section 3.2.5 in respect of voting rights of the Series A-1 Convertible Preferred Stock and Section 3.2.1(f) in respect of a Triggering Event, as such term is defined therein, and the formula to be applied in connection therewith), with respect to each of which requirements the Company has already been in compliance. The filing of the Series A-1 Certificate of Designation was unanimously approved by the Board of Directors on June 18, 2019. The affirmative approval of a majority of the holders of the Series A Convertible Preferred Stock for the exchange of such shares into shares of Series A-1 Convertible Preferred Stock occurred on or about June 19, 2019. The three holders of our Series A Convertible Preferred Stock were deemed to have exchanged their shares of Series A Convertible Preferred Stock for an equivalent number of shares of Series A-1 Convertible Preferred Stock, or an aggregate of 259,729 shares.
Except as described above, the rights, characteristics, and attributes of the Series A-1 Preferred Stock are the same as described below. Except as described above and as set forth below, references below to “Series A Preferred Stock” include and shall be deemed to refer to “Series A-1 Preferred Stock” on and after June 19, 2019.
Dividends
We cannot declare, pay or set aside any dividends on shares of any other class or series of our capital stock unless (in addition to the obtaining of any consents required by our Articles of Incorporation) the holders of the Series A Preferred Stock then outstanding shall first receive, or simultaneously receive, a dividend in the aggregate amount of $1.00, regardless of the number of then-issued and outstanding shares of Series A Preferred Stock. Any remaining dividends allocated by the Board of Directors shall be distributed in an equal amount per share to the holders of outstanding common stock and Series A Preferred Stock (on an as-if-converted to common stock basis pursuant to the Conversion Ratio as defined below).
Conversion
The Series A-1 Preferred Stock is not convertible into shares of our common stock or any other debt or equity securities of the Company. Further, the Series A-1 Preferred Stock is not convertible into shares of GeoTraq common stock except as described below.
The “Conversion Ratio” per share of the Series A-1 Preferred Stock in connection with any conversion shall be at a ratio of 458.3453:1, meaning every four hundred fifty-eight and 3,453/10,000ths (458.3453) shares of Series A-1 Preferred Stock, if and when converted into shares of GeoTraq common stock, shall convert into one share of GeoTraq common stock. Each holder of Series A-1 Preferred Stock shall have the right, exercisable at any time and from time to time (unless otherwise prohibited by law, rule, or regulation, or as restricted below), to convert any or all of such holder’s shares of Series A-1 Preferred Stock into shares of GeoTraq common stock at the Conversion Ratio.
Voting Rights
Each holder of a share of Series A Preferred Stock has a number of votes as is determined by multiplying (i) the number of shares of Series A Preferred Stock held by such holder, and (ii) 17. The holders of Series A-1 Preferred Stock vote together with all other classes and series of common and preferred stock of the Company as a single class on all actions to be taken by the common stockholders of the Company, except to the extent that voting as a separate class or series is required by law.
Redemption
The Series A-1 Preferred Stock has no redemption rights by JanOne, GeoTraq, or any other entity.
F-25
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Preemptive Rights
Holders of the Series A-1 Preferred Stock and holders of JanOne common stock are not entitled to any preemptive, subscription, or similar rights in respect of any securities of JanOne or GeoTraq, except as set forth in the Amended and Restated Series A-1 Certificate of Designation or in any other document agreed to by JanOne.
Protective Provisions
Without first obtaining the affirmative approval of a majority of the holders of the shares of Series A-1 Preferred Stock, we may not directly or indirectly (i) increase or decrease (other than by redemption or conversion) the total number of authorized shares of Series A-1 Preferred Stock or the total number of authorized or issued and outstanding shares of GeoTraq common stock; (ii) effect an exchange, reclassification, or cancellation of all or a part of the Series A-1 Preferred Stock, but excluding a stock split or reverse stock split or combination of the common stock or preferred stock; (iii) effect an exchange, or create a right of exchange, of all or part of the shares of another class of shares into shares of Series A-1 Preferred Stock; or (iii) alter or change the rights, preferences or privileges of the shares of Series A-1 Preferred Stock so as to affect adversely the shares of such series, including the rights set forth in this Designation; provided, however, that we may, without any vote of the holders of shares of the Series A-1 Preferred Stock, make technical, corrective, administrative or similar changes to the Amended and Restated Series A-1 Certificate of Designation that do not, individually or in the aggregate, materially adversely affect the rights or preferences of the holders of shares of the Series A-1 Preferred Stock.
|
Note 17: |
Stockholders’ Equity |
Common Stock: Our Articles of Incorporation authorize 200,000,000 shares of common stock that may be issued from time to time having such rights, powers, preferences and designations as the Board of Directors may determine.
On November 4, 2020, at the Company’s Annual Meeting of Stockholders (the “Annual Meeting”), the Company’s stockholders approved an amendment (the “Charter Amendment”) to the Company’s Articles of Incorporation to increase the total number of authorized shares of the Company’s common stock from 10,000,000 shares to 200,000,000 shares. Following stockholder approval, the Charter Amendment was filed with the Nevada Secretary of State on November 5, 2020, at which time the Charter Amendment became effective. The Company’s Board of Directors had previously approved the Charter Amendment, subject to stockholder approval. The description of the Charter Amendment is qualified in its entirety by reference to the complete text of the Charter Amendment, a copy of which is filed as Exhibit 3.10 to the Company’s Quarterly Report on Form 10-Q for the period ended September 26, 2020 and is incorporated herein by reference.
During fiscal year 2020, 104,798 shares of common stock were granted and issued in lieu of professional services at a fair value of $351. During fiscal year 2019, 224,483 common shares were issued as compensation to employees and consultants with a fair value of $500. Additionally, the Company recognized stock compensation expense of $54 and $131 during the years ended January 2, 2021 and December 31, 2019, respectively, related to the amortization of the fair value of shares of its common stock vesting and/or issued for services rendered.
As of January 2, 2021, and December 28, 2019, there were 1,829,982 and 1,919,048 shares, respectively, of common stock issued and outstanding.
Stock options: The 2016 Plan, which replaces the 2011 Plan, authorizes the granting of awards in any of the following forms: (i) incentive stock options, (ii) nonqualified stock options, (iii) restricted stock awards, and (iv) restricted stock units, and expires on the earlier of October 28, 2026, or the date that all shares reserved under the 2016 Plan are issued or no longer available. On November 4, 2020, the Company amended the 2016 Plan to increase the issuance of common shares from 400,000 to 800,000. The vesting period is determined by the Board of Directors at the time of the stock option grant. As of January 2, 2021 and December 28, 2019, 78,000 and 4,000, respectively, options were outstanding under the 2016 Plan.
Our 2011 Plan authorizes the granting of awards in any of the following forms: (i) stock options, (ii) stock appreciation rights, and (iii) other share-based awards, including but not limited to, restricted stock, restricted stock
F-26
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
units or performance shares, and expires on the earlier of May 12, 2021, or the date that all shares reserved under the 2011 Plan are issued or no longer available. As of January 2, 2021 and December 28, 2019, 35,900 and 40,400 options, respectively, were outstanding under the 2011 Plan. No additional awards will be granted under the 2011 Plan.
The following table summarizes stock option activity for fiscal 2020 and 2019:
|
|
|
Options |
|
|
Weighted Average Exercise |
|
|
Aggregate Intrinsic |
|
|
Weighted Average Remaining Contractual |
|
||||
|
|
|
Outstanding |
|
|
Price |
|
|
Value |
|
|
Life |
|
||||
|
Outstanding at December 29, 2018 |
|
|
100,900 |
|
|
$ |
11.07 |
|
|
$ |
— |
|
|
|
3.8 |
|
|
Cancelled/expired |
|
|
(56,500 |
) |
|
|
9.30 |
|
|
|
|
|
|
|
|
|
|
Outstanding at December 28, 2019 |
|
|
44,400 |
|
|
$ |
13.31 |
|
|
$ |
— |
|
|
|
3.0 |
|
|
Cancelled/expired |
|
|
(4,500 |
) |
|
|
9.45 |
|
|
|
|
|
|
|
|
|
|
Granted |
|
|
74,000 |
|
|
|
3.84 |
|
|
|
|
|
|
|
|
|
|
Outstanding at January 2, 2021 |
|
|
113,900 |
|
|
$ |
11.97 |
|
|
$ |
78 |
|
|
|
7.0 |
|
|
Exercisable at January 2, 2021 |
|
|
65,400 |
|
|
$ |
12.01 |
|
|
$ |
27 |
|
|
|
3.0 |
|
The exercise price for stock options outstanding and exercisable outstanding at January 2, 2021 is as follows:
|
Outstanding |
|
Exercisable |
||||||||
|
Number of Options |
|
|
Exercise Price ($) |
|
Number of Options |
|
|
Exercise Price ($) |
||
|
|
16,500 |
|
|
$17.35 to $23.45 |
|
|
16,500 |
|
|
$17.35 to $23.45 |
|
|
5,400 |
|
|
$11.10 to $15.00 |
|
|
5,400 |
|
|
$11.10 to $15.00 |
|
|
12,000 |
|
|
$5.70 to $9.90 |
|
|
12,000 |
|
|
$5.70 to $9.90 |
|
|
80,000 |
|
|
$3.54 to $5.25 |
|
|
31,500 |
|
|
$3.54 to $5.25 |
|
|
113,900 |
|
|
|
|
|
65,400 |
|
|
|
The following table summarizes information about the Company’s non-vested shares outstanding as of January 2, 2021 and December 28, 2019:
|
Non-vested Shares |
|
Number of Shares |
|
|
Average Grant-Date Fair Value |
|
||
|
Non-vested at December 29, 2018 |
|
|
— |
|
|
$ |
— |
|
|
Granted |
|
|
— |
|
|
$ |
— |
|
|
Vested |
|
|
— |
|
|
$ |
— |
|
|
Non-vested at December 28, 2019 |
|
|
— |
|
|
$ |
— |
|
|
Granted |
|
|
74,000 |
|
|
$ |
3.09 |
|
|
Vested |
|
|
(25,500 |
) |
|
$ |
3.85 |
|
|
Non-vested at January 2, 2021 |
|
|
48,500 |
|
|
$ |
3.83 |
|
We recognized share-based compensation expense related to stock options of $173 and $nil for fiscal years 2020 and 2019, respectively. At January 2, 2021, the Company has $55 of unrecognized compensation expense (net of estimated forfeitures) associated with stock option awards which the company expects to recognize as compensation expense through December 2021.
Warrants:
As of January 2, 2021 and December 28, 2019, there were 33,363 warrants outstanding to purchase 33,363 shares of common stock at a price of $3.40 per share that expire in November 2021.
F-27
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
|
Note 18: |
Loss per share |
Net loss per share is calculated using the weighted average number of shares of common stock outstanding during the applicable period. Basic weighted average common shares outstanding do not include shares of restricted stock that have not yet vested, although such shares are included as outstanding shares in the Company’s Consolidated Balance Sheet. Diluted net earnings per share is computed using the weighted average number of common shares outstanding and if dilutive, potential common shares outstanding during the period. Potential common shares consist of the additional common shares issuable in respect of restricted share awards, stock options and convertible preferred stock.
The following table presents the computation of basic and diluted net loss per share:
|
|
|
Fiscal Years Ended |
|
|||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Net loss |
|
$ |
(8,498 |
) |
|
$ |
(11,964 |
) |
|
Basic and diluted loss per share |
|
$ |
(4.59 |
) |
|
$ |
(6.78 |
) |
|
Weighted average common shares outstanding, basic and diluted |
|
|
1,852,147 |
|
|
|
1,763,670 |
|
Potentially dilutive securities totalling 406,992 and 337,492, respectively, were excluded from the calculation of diluted net loss per share for the years ended January 2, 2021 and December 28, 2019 because the effects were anti-dilutive based on the application of the treasury stock method.
|
Note 19: |
Major customers and suppliers |
For the fiscal year ended January 2, 2021, two customers represented 27% of our total revenues. For the fiscal year ended December 28, 2019, one customer represented 13% of our total revenues. As of January 2, 2021, three customers each represented 10% or more of our total trade receivables for a combined total of 37%. As of December 28, 2019, three customers, each represented more than 10% of our total trade receivables, for a total of 38% of our total trade receivables.
We purchased appliances for resale from four suppliers. We are continuing to secure other vendors from which to purchase appliances. However, the curtailment or loss of one of these suppliers or any appliance supplier could adversely affect our operations.
|
Note 20: |
Defined contribution plan |
We have a defined contribution salary deferral plan covering substantially all employees under Section 401(k) of the Internal Revenue Code of 1986, as amended (the “Code”). We contribute an amount equal to 10 cents for each dollar contributed by each employee up to a maximum of 5% of each employee’s compensation. We recognized expense for contributions to the plans of $20 and $61 for fiscal years 2020 and 2019, respectively.
|
Note 21: |
Segment information |
We operate within targeted markets through three reportable segments: biotechnology, recycling and technology. The biotechnology segment commenced operations in September 2019 and is focused on development of new and innovative solutions for ending the opioid epidemic ranging from digital technologies to educational advocacy. The recycling segment includes all fees charged and costs incurred for collecting, recycling and installing appliances for utilities and other customers. The recycling segment also includes byproduct revenue, which is primarily generated through the recycling of appliances. The nature of products, services and customers for both segments varies significantly. As such, the segments are managed separately. Our Chief Executive Officer has been identified as the Chief Operating Decision Maker (“CODM”). The CODM evaluates performance and allocates resources based on sales and income from operations of each segment. Operating loss represents revenues less cost of revenues and operating expenses, including certain allocated selling, general and administrative costs. There are no intersegment sales or transfers.
F-28
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
The following tables present our segment information for fiscal years 2020 and 2019:
|
|
|
Fiscal Years Ended |
|
|||||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Revenues |
|
|
|
|
|
|
|
|
|
Biotechnology |
|
$ |
— |
|
|
$ |
— |
|
|
Recycling |
|
|
33,867 |
|
|
|
35,097 |
|
|
Technology |
|
|
— |
|
|
|
— |
|
|
Total Revenues |
|
$ |
33,867 |
|
|
$ |
35,097 |
|
|
Gross profit |
|
|
|
|
|
|
|
|
|
Biotechnology |
|
$ |
— |
|
|
$ |
— |
|
|
Recycling |
|
|
8,827 |
|
|
|
7,786 |
|
|
Technology |
|
|
— |
|
|
|
— |
|
|
Total Gross profit |
|
$ |
8,827 |
|
|
$ |
7,786 |
|
|
Operating loss |
|
|
|
|
|
|
|
|
|
Biotechnology |
|
$ |
(1,738 |
) |
|
$ |
(1,038 |
) |
|
Recycling |
|
|
(3,172 |
) |
|
|
(6,397 |
) |
|
Technology |
|
|
(4,086 |
) |
|
|
(4,996 |
) |
|
Total Operating loss |
|
$ |
(8,996 |
) |
|
$ |
(12,431 |
) |
|
Depreciation and amortization |
|
|
|
|
|
|
|
|
|
Biotechnology |
|
$ |
— |
|
|
$ |
— |
|
|
Recycling |
|
|
376 |
|
|
|
346 |
|
|
Technology |
|
|
3,746 |
|
|
|
3,730 |
|
|
Total Depreciation and amortization |
|
$ |
4,122 |
|
|
$ |
4,076 |
|
|
Interest expense, net |
|
|
|
|
|
|
|
|
|
Biotechnology |
|
$ |
— |
|
|
$ |
— |
|
|
Recycling |
|
|
504 |
|
|
|
1,480 |
|
|
Technology |
|
|
— |
|
|
|
— |
|
|
Total Interest expense |
|
$ |
504 |
|
|
$ |
1,480 |
|
|
Net loss before provision for income taxes |
|
|
|
|
|
|
|
|
|
Biotechnology |
|
$ |
(1,738 |
) |
|
$ |
(1,038 |
) |
|
Recycling |
|
|
(2,980 |
) |
|
|
(9,008 |
) |
|
Technology |
|
|
(4,207 |
) |
|
|
(5,115 |
) |
|
Total Net loss before provision for income taxes |
|
$ |
(8,925 |
) |
|
$ |
(15,161 |
) |
|
|
|
As of |
|
|
As of |
|
||
|
|
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
|
Biotechnology |
|
$ |
— |
|
|
$ |
— |
|
|
Recycling |
|
|
10,614 |
|
|
|
11,505 |
|
|
Technology |
|
|
13,737 |
|
|
|
17,529 |
|
|
Total Assets |
|
$ |
24,351 |
|
|
$ |
29,034 |
|
|
Intangible Assets |
|
|
|
|
|
|
|
|
|
Biotechnology |
|
$ |
— |
|
|
$ |
— |
|
|
Recycling |
|
|
470 |
|
|
|
465 |
|
|
Technology |
|
|
13,519 |
|
|
|
17,240 |
|
|
Total Intangible Assets |
|
$ |
13,989 |
|
|
$ |
17,705 |
|
F-29
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
|
Note 22: |
Related parties |
Tony Isaac, the Company’s Chief Executive Officer, is the father of Jon Isaac, President and Chief Executive Officer of Live Ventures Incorporated (“Live Ventures”) and managing member of ICG, a greater than 5% stockholder of the Company. Tony Isaac, Chief Executive Officer, Virland Johnson, Chief Financial Officer, and Richard Butler, Board of Directors member of the Company, are Board of Directors member, Chief Financial Officer, and Board of Directors member, respectively, of Live Ventures. The Company also shares certain executive, accounting and legal services with Live Ventures. The total services shared were $243 and $193 for fiscal years ending January 2, 2021 and December 28, 2019, respectively. Connexx rents approximately 9,900 square feet of office space from Live Ventures at its Las Vegas, Nevada office. The total rent and common area expense for Connexx at the Las Vegas, Nevada office were $196 and $177 for fiscal years ending January 2, 2021 and December 28, 2019, respectively.
ApplianceSmart Note
On December 30, 2017, Purchaser entered into the Agreement with the Company and ApplianceSmart. Pursuant to the Agreement, the Purchaser purchased from the Company all of the Stock of ApplianceSmart in exchange for the Purchase Price. Effective April 1, 2018, the Purchaser issued the ApplianceSmart Note with a three-year term in the original principal amount of $3,919 for the balance of the purchase price. ApplianceSmart is guaranteeing the repayment of the ApplianceSmart Note.
On December 26, 2018, the ApplianceSmart Note was amended and restated to grant the Company a security interest in the assets of the Purchaser, ApplianceSmart, and ApplianceSmart Contracting Inc. in exchange for modifying the repayment terms to provide for the payment in full of all accrued interest and principal on April 1, 2021, the maturity date of the ApplianceSmart Note.
On March 15, 2019, the Company entered into subordination agreements with third parties pursuant to which it agreed to subordinate the payment of indebtedness under the ApplianceSmart Note and the Company’s security interest in the assets of ApplianceSmart and other related parties in exchange for up to $1,200 payable within 15 days of the agreement. ApplianceSmart can re-borrow up to the principal amount of the Note, $3,919. Additionally, the Company advanced ApplianceSmart $355 during fiscal 2019 under the ApplianceSmart Note.
On December 9, 2019, ApplianceSmart filed a voluntary petition (the “Chapter 11 Case”) in the United States Bankruptcy Court for the Southern District of New York (the “Bankruptcy Court”) seeking relief under Chapter 11 of Title 11 of the United States Code (the “Bankruptcy Code”).
For discussion related to potential obligations and or guarantees under ApplianceSmart Leases, see Note 15.
Related Party Note
On August 28, 2019 (amended August 25, 2020), ARCA Recycling entered into and delivered to ICG a secured revolving line of credit promissory note, whereby ICG agreed to provide the ARCA Recycling with a $2,500 revolving credit facility (the “ICG Note”). The Revolving Credit Facility matured on December 31, 2020. On March 30, 2021, ARCA Recycling entered into a Second Amendment and Waiver (the “Second Amendment”) to ICG Note. The Second Amendment extends the maturity date of the ICG Note from December 31, 2020 to August 18, 2021 and waives an event of default that occurred under the ICG Note (see Note 23 for complete discussion). ICG has not exercised its remedies or accelerated the indebtedness. The ICG Note bears interest at 8.75% per annum and provides for the payment of interest, monthly in arrears. ARCA Recycling pays a loan fee of 2.0% on each borrowing made under the ICG Note. In connection with entering into the Revolving Credit Facility, the Borrower also entered into a security agreement in favor of ICG, pursuant to which ARCA Recycling granted a security interest in all of its assets to ICG. The obligations of ARCA Recycling under the ICG Note are guaranteed by the Company. The foregoing transaction did not include the issuance of any shares of the Company’s common stock, warrants, or other derivative securities. ICG is a stockholder of the Company. Jon Isaac is the manager and sole member of ICG, and the son of Tony Isaac, the Chief Executive Officer of the Company and ARCA Recycling.
F-30
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
|
Note 23: |
Subsequent events |
The Company evaluated subsequent events through March 30, 2021, noting the following.
Equity Offering
On January 29, 2021, the Company entered into a Securities Purchase Agreement (the “Purchase Agreement”) with certain institutional investors (the “Purchasers”) for the sale by the Company in a registered direct offering (the “Offering”) of 571,428 shares of the Company’s common stock, par value $0.001 per share (the “Common Stock”), at a purchase price per share of Common Stock of $10.50.
On February 2, 2021, the Company closed its registered direct offering (the “Offering”) of an aggregate of 571,428 shares of Common Stock, at a price of $10.50 per share, for gross proceeds to the Company of approximately $6,000, before deducting placement agent fees and other offering expenses. The Company is utilizing the net proceeds for general working capital.
The Purchase Agreement contains customary representations, warranties and agreements by the Company and the Purchasers and customary indemnification rights and obligations of the parties. Pursuant to the terms of the Purchase Agreement, the Company has agreed to certain restrictions on the issuance and sale of its shares of Common Stock or Common Stock Equivalents (as defined in the Purchase Agreement) during the 45-day period following the closing of the Offering.
A.G.P./Alliance Global Partners acted as the sole placement agent (the “Placement Agent”) for the Company on a “reasonable best efforts” basis in connection with the Offering. The Company entered into a Placement Agency Agreement, dated as of January 29, 2021, by and between the Company and the Placement Agent (the “Placement Agency Agreement”). Pursuant to the Placement Agency Agreement, the Placement Agent will be entitled to a cash fee of 7% of the gross proceeds paid to the Company for the securities, reimbursement for accountable legal expenses incurred by it in connection with the Offering of up to $35.
The shares of Common Stock sold in the Offering were offered and sold by the Company pursuant to an effective shelf registration statement on Form S-3 (File No. 333-251645) (the “Registration Statement”), which was initially filed with the Securities and Exchange Commission on December 23, 2020, and was declared effective on December 29, 2020.
The representations, warranties and covenants contained in the Purchase Agreement were made solely for the benefit of the parties to the Purchase Agreement. In addition, such representations, warranties, and covenants (i) are intended as a way of allocating the risk between the parties to the Purchase Agreement and not as statements of fact, and (ii) may apply standards of materiality in a way that is different from what may be viewed as material by stockholders of, or other investors in, the Company. Accordingly, the Purchase Agreement is included with this filing only to provide investors with information regarding the terms of the transaction, and not to provide investors with any other factual information regarding the Company. Stockholders should not rely on the representations, warranties, and covenants or any descriptions thereof as characterizations of the actual state of facts or condition of the Company or any of its subsidiaries or affiliates. Moreover, information concerning the subject matter of the representations and warranties may change after the date of the Purchase Agreement, which subsequent information may or may not be fully reflected in public disclosures.
The foregoing descriptions of the Purchase Agreement and the Placement Agency Agreement are not complete and are qualified in their entireties by reference to the full text of the Purchase Agreement and the Placement Agency Agreement, a copy of each of which is filed as Exhibit 10.1 and Exhibit 1.1, respectively, to the Company’s Current Report on Form 8-K as field on January 29, 2021 and each is incorporated by reference herein.
F-31
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Sale of ARCA and Connexx
On February 19, 2021, the Company, together with its subsidiaries (a) ARCA Recycling, Inc., a California corporation (“ARCA”), and (b) Customer Connexx LLC, a Nevada limited liability company (“Connexx”), entered into an Asset Purchase Agreement (the “Purchase Agreement”) with (i) ARCA Affiliated Holdings Corporation, a Delaware corporation, (ii) ARCA Services Inc., a Delaware corporation, and (iii) Connexx Services Inc, a Delaware corporation (collectively, the “Buyers”), pursuant to which the Buyers agreed to acquire substantially all of the assets, and assume certain liabilities, of ARCA and Connexx (the “Disposition Transaction”). The principal of the Buyers is Virland A. Johnson, our Chief Financial Officer. The Disposition Transaction is expected to be consummated on or before August 18, 2021. In the event the Disposition Transaction is not closed by such date, the Purchase Agreement may be terminated and, in accordance with its terms, the Buyers may be required to pay to us a “break fee” of $250. The Purchase Agreement and the Disposition Transaction were unanimously approved by our Board of Directors at a meeting during the portion of which the Purchase Agreement and Disposition Transaction were considered and voted on Mr. Johnson was not present.
The purchase price that the Buyers have agreed to pay to us in the Disposition Transaction is $25,000, subject to certain adjustments, including a potential increase in the purchase price due to an earnout, the assumption of certain debt of ARCA, Connexx, or us, and potential indemnification claims (collectively, the “Initial Aggregate Consideration”). At closing, $7,500 of the Aggregate Consideration will be paid in immediately available funds, and $17,500 of the Initial Aggregate Consideration will be paid pursuant to the terms of the Buyers’ promissory note in our favor (the “Note”), which Note will bear interest at the rate of 6% per annum on the unpaid balance thereof. The Buyers’ payment obligations under the Note will be subordinated to the Buyers’ obligations to their Disposition Transaction lender(s), with the terms of such subordination to be determined upon Buyers’ identification of their lender(s). The parties have made customary representations, warranties, covenants, and indemnities in connection with the Disposition Transaction.
Commencing on February 19, 2021, (i) the Buyers will seek financing for the balance of the Initial Aggregate Consideration and (ii) the parties will prepare and negotiate the terms and conditions of certain ancillary documentation, including, without limitation, disclosure schedules, bills of sale, assignment and assumption agreements, the Note, and any related subordination documentation with Buyers’ Disposition Transaction lender(s).
The Purchase Agreement contains certain representations and warranties that the parties made to each other as of the date of the Purchase Agreement or such other date as specifically referenced therein. The representations and warranties were made solely for purposes of the Purchase Agreement and (i) are subject to limitations agreed by the parties in negotiating the terms and conditions thereof, (ii) may not be accurate or complete as of any specified date, (iii) will be qualified by the underlying disclosure schedules, (iv) may be subject to a contractual standard of materiality different from those generally applicable to investors, and (v) may have been used for the purpose of allocating risk among the parties thereto, rather than for establishing any matters as facts. Information concerning the subject matter of the representations and warranties may change after February 19, 2021, which subsequent information may or may not be fully reflected in JanOne’s public disclosures. For the foregoing reasons, the representations and warranties contained in the Purchase Agreement should not be relied upon as statements of factual information.
The foregoing descriptions of the Purchase Agreement and the Disposition Transaction do not purport to be complete and are qualified in their entirety by reference to the Purchase Agreement, a copy of which is filed with the Current Report on Form 8‑K as filed on February 25, 2021 as Exhibit 10.1 and is incorporated herein by reference.
Equipment Financing Agreement
On March 25, 2021, ARCA Recycling entered into a Master Equipment Finance Agreement (collectively, the “Equipment Finance Agreement”) with KLC Financial, Inc. (“KLC”). Under the terms of the Equipment Finance Agreement, KLC has agreed to make loans to ARCA Recycling secured by certain equipment purchased or to be purchased by ARCA Recycling on terms set forth or to be set forth in schedules to the Equipment Finance Agreement. Under the terms of Schedule No. 01 (the “Initial Loan”), KLC has loaned ARCA Recycling approximately $1.6 million secured by existing equipment of and new equipment to be purchased by ARCA
F-32
JANONE INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Dollars in thousands except per share amounts)
Recycling. ARCA Recycling will make monthly payments of $27 over a period of five years from the March 24, 2021, at which time it is intended that the Initial Loan will be repaid in full. The Initial Loan bears interest at 7.757% % per annum. KLC will have a first priority security interest over, among other things, all equipment identified in the schedules. The Initial Loan is guaranteed by Virland Johnson, the Chief Financial Officer of JanOne and Chief Financial Officer and Secretary of ARCA Recycling. The Equipment Finance Agreement contains customary affirmative and negative covenants, representations and warranties, and events of default for transactions of this nature. The foregoing description of the Equipment Finance Agreement does not purport to be complete and is qualified in its entirety by reference to the Equipment Finance Agreement, a copy of which is attached hereto as Exhibit 10.20 and is incorporated herein by reference.
Related Party Note
On March 30, 2021, ARCA Recycling entered into a Second Amendment and Waiver (the “Second Amendment”) to Secured Revolving Line of Credit Promissory Note (the “ICG Note”) with ICG. The Second Amendment extends the maturity date of the ICG Note from December 31, 2020 to August 18, 2021 and waives an event of default that occurred under the ICG Note. ICG is a record and beneficial owner of approximately 16% of the outstanding common stock of the Company. Jon Isaac is the manager and sole member of ICG, and the son of Tony Isaac, the Chief Executive Officer of JanOne and ARCA Recycling. The foregoing description of the Second Amendment does not purport to be complete and is qualified in its entirety by reference to the complete text of the Amendment, a copy of which is attached hereto as Exhibit 10.12 and is incorporated herein by reference.
F-33
ITEM 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosures
None.
ITEM 9A. Controls and Procedures
Evaluation of Disclosure control and Procedures. We carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of our disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)). Based upon that evaluation, our principal executive officer and principal financial officer concluded that, as of January 2, 2021, the period covered in this report, our disclosure controls and procedures were not effective to ensure that information required to be disclosed in reports filed under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the required time periods and is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure.
Changes in Internal Control Over Financial Reporting. There were no changes in the Company’s internal control over financial reporting during the quarter ended January 2, 2021, that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
Management’s Report on Internal Control Over Financial Reporting. Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)). Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of January 2, 2021. In making this assessment, we used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in 2013 regarding Internal Control – Integrated Framework. Based on our assessment using those criteria, our management concluded that our internal control over financial reporting was not effective as of January 2, 2021.
Management noted material weaknesses in internal control when conducting their evaluation of internal control as of January 2, 2021. (1) Insufficient information technology general controls (“ITGC”) and segregation of duties. It was noted that people who were negotiating a contract, were also involved in approving invoices without proper oversight. Additional controls and procedures are necessary and are being implemented to have check and balance on significant transactions and governance with those charged with governance authority. (2) Inadequate control design or lack of sufficient controls over significant accounting processes. The cutoff and reconciliation procedures were not effective with certain accrued and deferred expenses. (3) Insufficient assessment of the impact of potentially significant transactions, and (4) Insufficient processes and procedures related to proper recordkeeping of agreements and contracts. In addition, contract to invoice reconciliation was not effective with certain transportation service providers. As part of its remediation plan, processes and procedures have been implemented to help ensure accruals and invoices are reviewed for accuracy and properly recorded in the appropriate period. These material weaknesses remained outstanding as of the filing date of this annual report on Form 10-K and management is currently working to remedy these outstanding material weaknesses.
64
The Company’s management, including the Company’s CEO and CFO, do not expect that the Company’s disclosure controls and procedures or the Company’s internal control over financial reporting will prevent or detect all error and all fraud. A control system, regardless of how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system will be met. These inherent limitations include the following: judgements in decision-making can be faulty, and control and process breakdowns can occur because of simple errors or mistakes, controls can be circumvented by individuals, acting alone or in collusion with each other, or by management override, the design of any system of controls is based in part on certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected.
Item 1.01 Entry into a Material Definitive Agreement.
Related Party Note
On March 30, 2021, ARCA Recycling entered into a Second Amendment and Waiver (the “Second Amendment”) to Secured Revolving Line of Credit Promissory Note (the “ICG Note”) with ICG. The Second Amendment extends the maturity date of the ICG Note from December 31, 2020 to August 18, 2021 and waives an event of default that occurred under the ICG Note. ICG is a record and beneficial owner of approximately 16% of the outstanding common stock of the Company. Jon Isaac is the manager and sole member of ICG, and the son of Tony Isaac, the Chief Executive Officer of JanOne and ARCA Recycling. The foregoing description of the Second Amendment does not purport to be complete and is qualified in its entirety by reference to the complete text of the Amendment, a copy of which is attached hereto as Exhibit 10.12 and is incorporated herein by reference.
Equipment Financing Agreement
On March 25, 2021, ARCA Recycling entered into a Master Equipment Finance Agreement (collectively, the “Equipment Finance Agreement”) with KLC Financial, Inc. (“KLC”). Under the terms of the Equipment Finance Agreement, KLC has agreed to make loans to ARCA Recycling secured by certain equipment purchased or to be purchased by ARCA Recycling on terms set forth or to be set forth in schedules to the Equipment Finance Agreement. Under the terms of Schedule No. 01 (the “Initial Loan”), KLC has loaned ARCA Recycling approximately $1.6 million secured by existing equipment of and new equipment to be purchased by ARCA Recycling. ARCA Recycling will make monthly payments of $27 thousand over a period of five years from the March 24, 2021, at which time it is intended that the Initial Loan will be repaid in full. The Initial Loan bears interest at 7.757% % per annum. KLC will have a first priority security interest over, among other things, all equipment identified in the schedules. The Initial Loan is guaranteed by Virland Johnson, the Chief Financial Officer of JanOne and Chief Financial Officer and Secretary of ARCA Recycling. The Equipment Finance Agreement contains customary affirmative and negative covenants, representations and warranties, and events of default for transactions of this nature. The foregoing description of the Equipment Finance Agreement does not purport to be complete and is qualified in its entirety by reference to the Equipment Finance Agreement, a copy of which is attached hereto as Exhibit 10.20 and is incorporated herein by reference.
65
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The directors and executive officers of the Company and their ages as of January 2, 2021, are as follows:
|
Name |
|
Position with Company |
|
Age |
|
Richard D. Butler, Jr. |
|
Director |
|
71 |
|
Nael Hajjar |
|
Director |
|
36 |
|
John Bitar |
|
Director |
|
58 |
|
Tony Isaac |
|
President and Chief Executive Officer |
|
66 |
|
Virland A. Johnson |
|
Chief Financial Officer |
|
60 |
Richard D. Butler, Jr. has been a director of the Company since May 2015. Mr. Butler is the owner of an advisory firm that provides real estate, corporate, and financial advisory services since 1999, and is the co-Founder, Managing Director, and, since 2005, a major stockholder of Ref-Razzer Company, a whistle manufacturing and vending company. Prior to this, Mr. Butler was the Co-Founder and Executive Vice President of Aspen Healthcare, Inc. from 1996 to 1999. From 1993 to 1996, Mr. Butler was a Managing Director at Landmark Financial and from 1989 to 1993 he was a Partner at Cal Ventures Real Estate Investment Group. Prior to this, Mr. Butler has also served as the President and Chief Executive Officer of Mt. Whitney Savings Bank, Chief Executive Officer of First Federal Mortgage Bank, Chief Executive Officer of Trafalgar Mortgage, and Executive Officer and Member of the President’s Advisory Committee at State Savings & Loan Association (peak assets $14 billion) and American Savings & Loan Association (NYSE: FCA; peak assets $34 billion). Mr. Butler has served on the board of directors of Live Ventures (Nasdaq: LIVE) (“Live Ventures”) since August 2006. On December 9, 2019, ApplianceSmart, a subsidiary of Live Ventures, filed a voluntary petition in the United States Bankruptcy Court for the Southern District of New York seeking relief under Chapter 11 of Title 11 of the United States Code. Mr. Butler attended Bowling Green University in Ohio, San Joaquin Delta College in California, and Southern Oregon State College. We believe that Mr. Butler brings to the Board extensive experience in financial management and executive roles, which enable him to provide important expertise in financial, operating and strategic matters that impact our Company.
Nael Hajjar has been a director of the Company since August 2018. Mr. Hajjar is currently the Unit Head for the Annual Wholesale Trade Survey in Statistics Canada’s Manufacturing and Wholesale Trade Division. From March 2011 through May 2016, Mr. Hajjar was a Senior Analyst – Economist of Statistics Canada’s Producer Prices Division where he developed Canada’s first ever Investment Banking Services Price Index while leading the development of a variety of Financial Services Price Index development projects. We believe that Mr. Hajjar brings to the Board extensive experience in research and analysis of financial statistics, economics, and business practices in a variety of industries including manufacturing, logging, Wholesale Trade, and financial services. We believe that Mr. Hajjar also has extensive experience in project management, and he holds a Bachelor of Social Science, Honors in Economics, and Bachelor of Commerce, Option in Finance, from the University of Ottawa.
John Bitar has been a director of the Company since January 2020. Since 2012, Mr. Bitar has been providing consulting services to companies and clients on business and legal strategies, management, operations, and cost controls. From 2007 to 2012, Mr. Bitar co-founded and was Managing Partner of a worker’s compensation law firm. Mr. Bitar has been an attorney admitted to the California State Bar since 1999. Mr. Bitar graduated from the University of Southern California in 1996 and earned his Juris Doctorate Degree in 1999 from University of the Pacific, McGeorge School of Law. We believe that Mr. Bitar has significant business experience and brings operational expertise to the Board.
Tony Isaac has been a director of the Company since May 2015 and Chief Executive Officer of the Company since May 2016. He served as Interim Chief Executive Officer of the Company from February 2016 until May 2016. Mr. Isaac has served as Financial Planning and Strategist/Economist of Live Ventures since July 2012. He is the Chairman and Co-Founder of Isaac Organization, a privately held investment company. Mr. Isaac has invested in various companies, both private and public from 1980 to present. Mr. Isaac’s specialty is negotiation and problem-solving of complex real estate and business transactions. Mr. Isaac has served as a director of Live Ventures since December 2011. On December 9, 2019, ApplianceSmart, Inc. (“ApplianceSmart”), a subsidiary of Live Ventures, filed a voluntary petition in the United States Bankruptcy Court for the Southern District of New York seeking relief under Chapter 11 of Title 11 of the United States Code. Mr. Isaac graduated from Ottawa University in 1981, where he majored in Commerce and Business Administration and Economics. We believe that Mr. Isaac has significant investment and financial expertise and public board experience that he brings to the Board.
66
Virland A. Johnson was appointed Chief Financial Officer of the Company on August 21, 2017. Mr. Johnson had previously served the Company as a consultant beginning in February 2017. Mr. Johnson also continues to serve as Chief Financial Officer for Live Ventures Incorporated, a holding company of diversified businesses (Nasdaq: LIVE). On December 9, 2019, ApplianceSmart, a subsidiary of Live Ventures, filed a voluntary petition in the United States Bankruptcy Court for the Southern District of New York seeking relief under Chapter 11 of Title 11 of the United States Code. Mr. Johnson is a director and Chief Financial Officer and Secretary of ApplianceSmart. Prior to joining Live Ventures Incorporated, Mr. Johnson was Sr. Director of Revenue for JDA Software from February 2010 to April 2016, where he was responsible for revenue recognition determination, sales and contract support while acting as a subject matter expert. Prior to joining JDA, Mr. Johnson provided leadership and strategic direction while serving in C-Level executive roles in public and privately held companies such as Cultural Experiences Abroad, Inc., Fender Musical Instruments Corp., Triumph Group, Inc., Unitech Industries, Inc. and Younger Brothers Group, Inc. Mr. Johnson’s more than 25 years of experience is primarily in the areas of process improvement, complex debt financings, SEC and financial reporting, turn-arounds, corporate restructuring, global finance, merger and acquisitions and returning companies to profitability and enhancing stockholder value. Mr. Johnson holds a Bachelor’s degree in Accountancy from Arizona State University.
Delinquent Section 16(a) Reports
Section 16(a) of the Securities Exchange Act of 1934, as amended, requires the Company’s officers and directors, and persons who own more than 10% of a registered class of the Company’s equity securities, to file reports of ownership on Form 3 and changes in ownership on Form 4 or Form 5 with the SEC. Such officers, directors and 10% shareholders are also required by SEC rules to furnish the Company with copies of all Section 16(a) forms they file.
Based solely on its review of copies of such forms received by it, or written representations from certain reporting persons, the Company believes that, during the fiscal year ended January 2, 2021, except as noted below, all of its officers, directors and 10% stockholders timely complied with all Section 16(a) filing requirements: Form 4 filed late by Virland A. Johnson, the Company’s Chief Financial Officer, on September 3, 2020 with respect to several transactions that occurred in March 2020.
Code of Ethics
Our Audit Committee has adopted a code of ethics applicable to our directors and officers (including our Chief Executive Officer and Chief Financial Officer) and other of our senior executives and employees in accordance with applicable rules and regulations of the SEC and The Nasdaq Stock Market. A copy of the code of ethics may be obtained upon request, without charge, by addressing a request to Investor Relations, JanOne Inc., 325 E. Warm Springs Road, Suite 102, Las Vegas, Nevada 89119. The code of ethics is also posted on our website at www.janone.com under “Investor Relations — Corporate Governance.”
We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding the amendment to, or waiver from, a provision of the code of ethics by posting such information on our website at the address and location specified above and, to the extent required by the listing standards of the Nasdaq Capital Market, by filing a Current Report on Form 8-K with the SEC disclosing such information.
Audit Committee
The Audit Committee of the Board of Directors is comprised entirely of non-employee directors. In fiscal 2020, the members of the Audit Committee were Mr. Butler (Chair), Mr. Bitar, and Mr. Hajjar. Each of Messrs. Bitar, Butler, and Hajjar was an “independent” director as defined under Nasdaq rules. The Audit Committee is responsible for selecting and approving the Company’s independent auditors, for relations with the independent auditors, for review of internal auditing functions (whether formal or informal) and internal controls, and for review of financial reporting policies to assure full disclosure of financial condition. The Audit Committee operates under a written charter adopted by the Board of Directors, which is posted on the Company’s website at www.janone.com under the caption “Investor Relations - Governance.” The Board has determined that Mr. Butler is an “audit committee financial expert” as defined in SEC rules.
67
Compensation and Benefits Committee
The Compensation Committee of the Board of Directors is comprised entirely of non-employee directors. In fiscal 2020, the members of the Compensation Committee were Mr. Butler (Chair) and Mr. Hajjar, each of whom was also an “independent” director as defined under Nasdaq rules. The Compensation Committee is responsible for review and approval of officer salaries and other compensation and benefits programs and determination of officer bonuses. Annual compensation for the Company’s executive officers, other than the CEO, is recommended by the CEO and approved by the Compensation Committee. The annual compensation for the CEO is recommended by the Compensation Committee and formally approved by the full Board of Directors.
In the performance of its duties, the Compensation Committee may select independent compensation consultants to advise the committee when appropriate. In addition, the Compensation Committee may delegate authority to subcommittees where appropriate. The Compensation Committee may separately meet with management if deemed necessary and appropriate. The Compensation Committee operates under a written charter adopted by the Board of Directors in March 2011, which is posted on the Company’s website at www.janone.com under the caption “Investor Relations - Governance.”
Governance Committee
The Nominating and Corporate Governance Committee (the “Governance Committee”) is comprised entirely of non-employee directors. In fiscal 2020, the members of the Governance Committee were Mr. Butler (Chair) and Mr. Bitar, each of whom was also an “independent” director as defined under NASDAQ rules. The primary purpose of the Governance Committee is to ensure an appropriate and effective role for the Board of Directors in the governance of the Company. The principal recurring duties and responsibilities of the Governance Committee include (i) making recommendations to the Board regarding the size and composition of the Board, (ii) identifying and recommending to the Board of Directors candidates for election as directors, (iii) reviewing the Board’s committee structure, composition and membership and recommending to the Board candidates for appointment as members of the Board’s standing committees, (iv) reviewing and recommending to the Board corporate governance policies and procedures, (v) reviewing the Company’s Code of Business Ethics and Conduct and compliance therewith, and (vi) ensuring that emergency succession planning occurs for the positions of Chief Executive Officer, other key management positions, the Board chairperson and Board members. The Governance Committee operates under a written charter adopted by the Board of Directors in March 2011, which is posted on the Company’s website at www.janone.com under the caption “Investor Relations - Governance.”
The Governance Committee will consider director candidates recommended by shareholders. The criteria applied by the Governance Committee in the selection of director candidates is the same whether the candidate was recommended by a Board member, an executive officer, a shareholder or a third party, and accordingly, the Governance Committee has not deemed it necessary to adopt a formal policy regarding consideration of candidates recommended by shareholders. Shareholders wishing to recommend candidates for Board membership should submit the recommendations in writing to the Secretary of the Company.
The Governance Committee identifies director candidates primarily by considering recommendations made by directors, management and stockholders. The Governance Committee also has the authority to retain third parties to identify and evaluate director candidates and to approve any associated fees or expenses. Board candidates are evaluated on the basis of a number of factors, including the candidate’s background, skills, judgment, diversity, experience with companies of comparable complexity and size, the interplay of the candidate’s experience with the experience of other Board members, the candidate’s independence or lack of independence, and the candidate’s qualifications for committee membership. The Governance Committee does not assign any particular weighting or priority to any of these factors and considers each director candidate in the context of the current needs of the Board as a whole. Director candidates recommended by shareholders are evaluated in the same manner as candidates recommended by other persons.
68
ITEM 11. EXECUTIVE COMPENSATION
The following table sets forth the cash and non-cash compensation for fiscal years ended January 2, 2021 and December 28, 2019, earned by each person who served as Chief Executive Officer and our other two most highly compensated executive officers who held office as of January 2, 2021 (“named executive officers”):
Summary Compensation Table
|
Name and Principal Position (1) |
|
Year |
|
Salary ($) |
|
|
Bonus ($) |
|
|
Stock Award ($) |
|
|
|
Option Awards ($) |
|
|
All Other Compensation ($) |
|
|
Total ($) |
|
||||||
|
Tony Isaac |
|
2020 |
|
|
534,471 |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
534,471 |
|
|
President and Chief Executive Officer |
|
2019 |
|
|
571,427 |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
571,427 |
|
|
Eric Bolling (2) |
|
2020 |
|
|
301,442 |
|
|
|
— |
|
|
|
54,203 |
|
|
|
|
— |
|
|
|
— |
|
|
|
355,645 |
|
|
Former President |
|
2019 |
|
|
148,077 |
|
|
|
— |
|
|
|
500,000 |
|
|
|
|
— |
|
|
|
— |
|
|
|
648,077 |
|
|
Virland A. Johnson |
|
2020 |
|
|
121,731 |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
121,731 |
|
|
Chief Financial Officer |
|
2019 |
|
|
125,274 |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
125,274 |
|
|
(1) |
The Company only had two executive officers as of January 2, 2021. |
|
(2) |
On August 9, 2020, we entered into a first amendment to amendment and restated employment agreement (the “Employment Agreement Amendment”) with Eric Bolling. Under the terms of the Employment Agreement Amendment, in exchange for the Company issuing Mr. Bolling 40,000 shares of fully vested, restricted common stock of the Company (the “August 2020 Shares”), Mr. Bolling (i) agreed to continue to provide the services described in his employment agreement, (ii) resigned his position as President of the Company and Chairman of the Board of Directors (the “Board”), provided that Mr. Bolling will continue as a member of the Board and further agreed that it is in the Company’s sole discretion whether Mr. Bolling continues as a member of the Board following the Company’s 2020 Annual Meeting of Stockholders; (iii) agreed to forego his base salary on a going forward basis and further agreed that he is not entitled to any base salary or any further remuneration or compensation from the Company whatsoever (other than the August 2020 Shares) after August 1, 2020, and (iv) forfeited the other 70,607 shares of the Company’s common stock that was owed to him under the terms of the employment agreement prior to the execution and delivery of the Employment Agreement Amendment. This amount reflects the fair value of a stock grant awarded to Mr. Bolling, as discussed. |
Outstanding Equity Awards at January 2, 2021
The following table provides a summary of equity awards outstanding for our Named Executive Officers at January 2, 2021:
|
Name |
Number of Securities Underlying Unexercised Options (in shares) Exercisable |
|
|
Number of Securities Underlying Unexercised Options (in shares) Unexercisable |
|
|
Option Exercise Price ($) |
|
|
Option Expiration Date |
|||
|
Tony Isaac |
|
2,000 |
|
|
|
— |
|
|
|
5.25 |
|
|
05/18/2025 |
|
Eric Bolling (1) |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
Virland A. Johnson |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
(1) |
During August 2020, Mr. Bolling resigned his position as President of the Company and Chairman of the Board. |
Stock Option Plans
The Company uses stock options to attract and retain executives, directors, consultants and key employees. Stock options are currently outstanding under three stock option plans. The Company’s 2016 Equity Incentive Plan (the
69
“2016 Plan”) was adopted by the Board of Directors in October 2016 and approved by the shareholders at the 2016 annual meeting of shareholders. Under the 2016 Plan, the Company has reserved an aggregate of 400,000 shares of its common stock for option grants. On November 4, 2020, at the Annual Meeting, the Company’s stockholders approved an amendment (the “Plan Amendment”) to the 2016 Plan to increase the total number of shares of the Company’s common stock reserved for issuance under the 2016 Plan to from 400,000 shares to 800,000 shares. The Company’s 2011 Stock Compensation Plan (the “2011 Plan”) was adopted by the Board of Directors in March 2011 and approved by the shareholders at the 2011 annual meeting of shareholders. The 2011 Plan expired on December 29, 2016, but options granted under the 2011 Plan before it expired will continue to be exercisable in accordance with their terms. As of January 2, 2021, options to purchase an aggregate of 113,900 shares were outstanding, including options for 78,000 shares under the 2016 Plan and options for 35,900 shares under the 2011 Plan. The Plans are administered by the Compensation Committee or the full Board of Directors acting as the Committee.
The 2016 Plan permits the grant of the following types of awards, in the amounts and upon the terms determined by the Administrator:
|
|
• |
Options. Options may either be incentive stock options (“ISOs”) which are specifically designated as such for purposes of compliance with Section 422 of the Internal Revenue Code or non-qualified stock options (“NSOs”). Options shall vest as determined by the Administrator, subject to certain statutory limitations regarding the maximum term of ISOs and the maximum value of ISOs that may vest in one year. The exercise price of each share subject to an ISO will be equal to or greater than the fair market value of a share on the date of the grant of the ISO, except in the case of an ISO grant to a stockholder who owns more than 10% of the Company’s outstanding shares, in which case the exercise price will be equal to or greater than 110% of the fair market value of a share on the grant date. The exercise price of each share subject to an NSO shall be determined by the Board at the time of grant but will be equal to or greater than the fair market value of a share on the date of grant. Recipients of options have no rights as a stockholder with respect to any shares covered by the award until the award is exercised and a stock certificate or book entry evidencing such shares is issued or made, respectively. |
|
|
• |
Restricted Stock Awards. Restricted stock awards consist of shares granted to a participant that are subject to one or more risks of forfeiture. Restricted stock awards may be subject to risk of forfeiture based on the passage of time or the satisfaction of other criteria, such as continued employment or Company performance. Recipients of restricted stock awards are entitled to vote and receive dividends attributable to the shares underlying the award beginning on the grant date. |
|
|
• |
Restricted Stock Units. Restricted stock units consist of a right to receive shares (or cash, in the Administrator’s discretion) on one or more vesting dates in the future. The vesting dates may be based on the passage of time or the satisfaction of other criteria, such as continued employment or Company performance. Recipients of restricted stock units have no rights as a stockholder with respect to any shares covered by the award until the date a stock certificate or book entry evidencing such shares is issued or made, respectively. |
70
Compensation of Non-Employee Directors
The Company uses cash compensation to attract and retain qualified candidates to serve on the Board of Directors. In setting director compensation, the Company considers the significant amount of time that directors expend fulfilling their duties to the Company as well as the skill level required by the Company of members of the Board. All of the Company’s directors are reimbursed for reasonable travel expenses incurred in attending meetings.
The table below presents cash and non-cash compensation paid to non-employee directors during the last fiscal year.
Non-Management Director Compensation for Fiscal Year Ended January 2, 2021
|
Name |
|
Fees Earned or Paid in Cash ($) |
|
|
Option Awards ($) |
|
|
All Other Compensation ($) |
|
|
Total ($) |
|
||||
|
|
|
17,758 |
|
|
|
— |
|
|
|
— |
|
|
|
17,758 |
|
|
|
Richard D. Butler, Jr. |
|
|
30,000 |
|
|
|
— |
|
|
|
— |
|
|
|
30,000 |
|
|
Nael Hajjar |
|
|
14,400 |
|
|
|
— |
|
|
|
— |
|
|
|
14,400 |
|
71
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED SHAREHOLDER MATTERS
The following table sets forth as of March 25, 2021 the beneficial ownership of common stock by each of the Company’s directors, each of the named executive officers, and all directors and executive officers of the Company as a group, as well as information about beneficial owners of 5% or more of the Company’s voting securities. Beneficial ownership includes shares that may be acquired in the next 60 days through the exercise of options or warrants.
|
Beneficial Owner |
|
Position with Company |
|
Number of Shares Beneficially Owned (1) |
|
|
Percent of Outstanding Common (2) |
|
||
|
Directors and executive officers: |
|
|
|
|
|
|
|
|
|
|
|
Tony Isaac (3) |
|
President and Chief Executive Officer |
|
|
94,000 |
|
|
|
3.9 |
% |
|
Virland A. Johnson |
|
Chief Financial Officer |
|
|
32,933 |
|
|
|
1.4 |
% |
|
Richard D. Butler, Jr. (3) |
|
Director |
|
|
18,000 |
|
|
* |
|
|
|
John Bitar |
|
Director |
|
|
2,000 |
|
|
* |
|
|
|
Nael Hajjar |
|
Director |
|
|
— |
|
|
* |
|
|
|
All directors and executive officers as a group (6 persons) (3) |
|
|
|
|
146,933 |
|
|
|
6.1 |
% |
|
Other 5% shareholders: |
|
|
|
|
|
|
|
|
|
|
|
Isaac Capital Group, LLC (4) |
|
|
|
|
392,941 |
|
|
|
16.3 |
% |
|
Altium Capital Management (5) |
|
|
|
|
190,476 |
|
|
|
7.9 |
% |
|
Ionic Ventures, LLC (6) |
|
|
|
|
190,476 |
|
|
|
7.9 |
% |
|
* |
Indicates ownership of less than 1% of the outstanding shares |
|
(1) |
Unless otherwise noted, each person or group identified possesses sole voting and investment power with respect to such shares. |
|
(2) |
Applicable percentage of ownership is based on 2,403,410 shares of common stock outstanding as of March 25, 2020 plus, for each shareholder, all shares that such shareholder could purchase within 60 days upon the exercise of existing stock options. |
|
(3) |
Includes shares which could be purchased within 60 days upon the exercise of existing stock options or warrants, as follows: Mr. Isaac, 2,000 shares; Mr. Butler, 4,000 shares and Mr. Bitar 2,000 shares; and all directors and executive officers as a group, 8,000 shares. The address for each individual is 325 E. Warm Springs Road Suite 102, Las Vegas, Nevada, 89119. |
|
(4) |
According to a Schedule 13G filed April 30, 2019, Isaac Capital Group, LLC (“ICG”) beneficially owned 392,941 shares of common stock. ICG has sole dispositive power as to all 392,941 shares and sole voting power as to 392,941 shares. The address for ICG is 505 East Windmill Lane, Suite 1C-295, Las Vegas Nevada 89123. |
|
(5) |
According to a Schedule 13G filed February 8, 2021, Altium Growth Fund, LP (the “Fund”), Altium Capital Management, LP, and Altium Growth GP, LLC (collectively “Altium”), has shared voting power and dispositive power with respect to 190,476 shares of common stock. According to this Schedule 13G, the Fund is the record and direct beneficial owner of the securities covered by this statement. Altium Capital Management, LP is the investment adviser of, and may be deemed to beneficially own securities, owned by, the Fund. Altium Growth GP, LLC is the general partner of, and may be deemed to beneficially own securities owned by, the Fund. The Schedule 13G lists Altium’s principal place of business as 152 West 57 Street, FL 20, New York, NY 10019. |
(6) According to a Schedule 13G filed February 1, 2021, Ionic Ventures LLC (“Ionic Ventures”) beneficially owned 190,476 shares of common stock. Ionic Ventures is controlled by Brendan O’Neil and Keith Coulston and has a principal address of 3053 Fillmore St., Suite 256 San Francisco, CA 94123.
72
Beneficial Ownership of Series A Preferred Stock
The following table sets forth as of March 25, 2021 the beneficial ownership of Series A-1 Preferred Stock by each owner of 5% or more of the Company’s Series A-1 Preferred Stock. No officers or directors of the Company have beneficial ownership of Series A-1 Preferred Stock. There are no options or warrants to purchase shares of Series A-1 Preferred Stock.
|
Beneficial Owner |
|
Number of Shares Beneficially Owned (1) |
|
|
Percent of Outstanding Series A Preferred (2) |
|
||
|
Gregg Sullivan (3) |
|
|
28,859 |
|
|
|
11.1 |
% |
|
Juan Yunis (4) |
|
|
216,729 |
|
|
|
83.4 |
% |
|
Isaac Capital Group, LLC (5) |
|
|
14,141 |
|
|
|
5.5 |
% |
|
(1) |
Unless otherwise noted, each person or group identified possesses sole voting and investment power with respect to such shares. |
|
(2) |
Applicable percentage of ownership is based on 259,729 shares of Series A-1 Preferred Stock outstanding as of March 25, 2021. |
|
(3) |
The business address for Mr. Sullivan is c/o JanOne Inc., 325 E. Warm Springs Road, Suite 102, Las Vegas, Nevada 89119. On January 16, 2019, GeoTraq terminated the employment of Mr. Sullivan pursuant to the terms of the employment agreement dated August 18, 2017 (the “Employment Agreement”) between GeoTraq and Mr. Sullivan. Under the terms of the Employment Agreement, 28,859 of the shares of the Company’s Series A Preferred Stock owned by Mr. Sullivan immediately prior to the termination are deemed to have been returned to the Company’s treasury for cancellation effective as of January 16, 2019, without the requirement that either Mr. Sullivan or the Company take any further action. |
|
(4) |
The business address for Mr. Yunis solely with respect to the shares of Series A-1 Preferred Stock is c/o JanOne Inc., 325 E. Warm Springs Road, Suite 102, Las Vegas, Nevada 89119. |
|
(5) |
The address for Isaac Capital Group, LLC is 505 East Windmill Lane, Suite 1C-295, Las Vegas Nevada 89123. |
The following table provides aggregate information under our equity compensation plans as of January 2, 2021:
|
|
|
(a) |
|
|
(b) |
|
|
(c) |
|
|||
|
|
|
Number of Securities to be Issued Upon Exercise of Outstanding Options and Warrants |
|
|
Weighted Average Exercise Price of Outstanding Options, Warrants and Rights |
|
|
Number of Securities Available for Future Issuance Under Equity Compensation Plans, Excluding Securities Reflected in Column (a) |
|
|||
|
Equity compensation plans approved by stockholders |
|
|
113,900 |
|
|
$ |
11.97 |
|
|
|
780,000 |
|
|
Equity compensation plans not approved by stockholders |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Total |
|
|
113,900 |
|
|
$ |
11.97 |
|
|
|
780,000 |
|
73
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Review, Approval or Ratification of Transactions with Related Persons
There are no family relationships among any of the directors or executive officers of the Company. Of the current directors, each of Messrs. Butler, Bitar, and Hajjar is an “independent” director, as defined under the rules of The Nasdaq Stock Market (“Nasdaq”) and each has been an independent director since each joined the Board.
In accordance with its charter, the Audit Committee reviews and recommends for approval all related party transactions (as such term is defined for purposes of Item 404 of Regulation S-K). The Audit Committee participated in the approval of the transactions described above.
Related Party Transactions (stated in thousands of dollars)
Tony Isaac, the Company’s Chief Executive Officer, is the father of Jon Isaac, President and Chief Executive Officer of Live Ventures and managing member of ICG, a greater than 5% stockholder of the Company. Tony Isaac, Chief Executive Officer, Virland Johnson, Chief Financial Officer, Richard Butler, Board of Directors member, and Dennis Gao, Board of Directors member of the Company, are Board of Directors member, Chief Financial Officer, Board of Directors member, and Board of Directors members, respectively, of Live Ventures. The Company also shares certain executive, accounting and legal services with Live Ventures. The total services shared were $243 and $193 for fiscal years ending January 2, 2021 and December 28, 2019, respectively. Customer Connexx rents approximately 9,900 square feet of office space from Live Ventures at its Las Vegas, NV office. The total rent and common area expense were $196 and $177 for fiscal years ending January 2, 2021 and December 28, 2019, respectively.
ApplianceSmart Note
On December 30, 2017, Purchaser entered into the Agreement with the Company and ApplianceSmart. Pursuant to the Agreement, the Purchaser purchased from the Company all of the Stock of ApplianceSmart in exchange for the Purchase Price. Effective April 1, 2018, the Purchaser issued the ApplianceSmart Note with a three-year term in the original principal amount of $3,919 for the balance of the purchase price. ApplianceSmart is guaranteeing the repayment of the ApplianceSmart Note.
On December 26, 2018, the ApplianceSmart Note was amended and restated to grant the Company a security interest in the assets of the Purchaser, ApplianceSmart, and ApplianceSmart Contracting Inc. in exchange for modifying the repayment terms to provide for the payment in full of all accrued interest and principal on April 1, 2021, the maturity date of the ApplianceSmart Note.
On March 15, 2019, the Company entered into subordination agreements with third parties pursuant to which it agreed to subordinate the payment of indebtedness under the ApplianceSmart Note and the Company’s security interest in the assets of ApplianceSmart and other related parties in exchange for up to $1,200 payable within 15 days of the agreement. ApplianceSmart can re-borrow up to the principal amount of the Note of $3,919. Additionally, the Company advanced ApplianceSmart $355 during fiscal 2019 under the ApplianceSmart Note.
On December 9, 2019, ApplianceSmart filed a voluntary petition (the “Chapter 11 Case”) in the United States Bankruptcy Court for the Southern District of New York (the “Bankruptcy Court”) seeking relief under Chapter 11 of Title 11 of the United States Code (the “Bankruptcy Code”).
74
Related Party Note
On August 28, 2019 (amended August 25, 2020), ARCA Recycling entered into and delivered to ICG a secured revolving line of credit promissory note, whereby the Lender agreed to provide the ARCA Recycling with a $2,500 revolving credit facility (the “ICG Note”). The ICG Note matured on December 31, 2020. On March 30, 2021, ARCA Recycling entered into a Second Amendment and Waiver (the “Second Amendment”) to Secured Revolving Line of Credit Promissory Note (the “ICG Note”) with ICG. The Second Amendment extends the maturity date of the ICG Note from December 31, 2020 to August 18, 2021 and waives an event of default that occurred under the ICG Note. ICG has not exercised its remedies or accelerated the indebtedness. The ICG Note bears interest at 8.75% per annum and provides for the payment of interest, monthly in arrears. ARCA Recycling pays a loan fee of 2.0% on each borrowing made under the ICG Note. In connection with entering into the ICG Note, the Borrower also entered into a security agreement in favor of ICG, pursuant to which ARCA Recycling granted a security interest in all of its assets to ICG. The obligations of ARCA Recycling under the ICG Note are guaranteed by the Company. The foregoing transaction did not include the issuance of any shares of the Company’s common stock, warrants, or other derivative securities. ICG is a stockholder of the Company. Jon Isaac is the manager and sole member of ICG, and the son of Tony Isaac, the Chief Executive Officer of the Company and ARCA Recycling.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Each year, the Audit Committee approves the annual audit engagement in advance. The Audit Committee also has established procedures to pre-approve all non-audit services provided by the Company’s independent registered public accounting firm. All fiscal 2020 and 2019 non-audit services listed below were pre-approved.
Audit and Audit-Related Fees: This category includes the audit of our annual financial statements and review of financial statements included in our annual and periodic reports that are filed with the SEC. This category also includes services performed for the preparation of responses to SEC and Nasdaq correspondence, travel expenses for our auditors, on audit and accounting matters that arose during, or as a result of, the audit or the review of interim financial statements, and the preparation of an annual “management letter” on internal control and other matters.
Tax Fees: This category consists of professional services rendered by our independent auditors for tax compliance.
All Other Fees consist of fees for services other than the services described above.
The following fees were billed to us by our independent registered public accounting firm, WSRP, LLC (“WSRP”) for 2020 and WSRP and SingerLewak LLP for 2019. SingerLewak LLP served as the Company’s auditor from fiscal 2017 through October 2019 and reviewed the Company’s quarterly financial statements for each of the first two fiscal quarters during fiscal 2019. WSRP was appointed the Company’s auditor during October 2019.
|
Description |
|
January 2, 2021 |
|
|
December 28, 2019 |
|
||
|
Audit fees |
|
$ |
212,725 |
|
|
$ |
219,549 |
|
|
Audit-related fees |
|
|
11,466 |
|
|
|
— |
|
|
Tax fees |
|
|
48,459 |
|
|
|
79,201 |
|
|
All other fees |
|
|
— |
|
|
|
— |
|
|
Total |
|
$ |
272,650 |
|
|
$ |
298,750 |
|
75
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
|
(a) |
Financial Statements, Financial Statement Schedules and Exhibits |
|
|
1 |
Financial Statements |
See Index to Financial Statements under Item 8 of this report.
|
|
2 |
Financial Statement Schedules |
None.
|
|
3 |
Exhibits |
See Index to Exhibits
None.
76
|
Exhibit No. |
|
Description |
|
2.1 |
|
Agreement and Plan of Merger dated August 18, 2017, between the Company, Appliance Recycling Acquisition Corp., GeoTraq Inc., and the stockholders of GeoTraq Inc. [filed as Exhibit 10.9 to the Company’s Form 10-Q/A for the quarterly period ended July 1, 2017 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
2.2 |
|
Stock Purchase Agreement dated December 30, 2017 [filed as Exhibit 10.28 to the Company’s Form 10-K for the fiscal year ended December 30, 2017 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
2.3 |
|
Asset Purchase Agreement among JanOne Inc., ARCA Recycling, Inc., and Customer Connexx LLC, on the one hand, and ARCA Affiliated Holdings Corporation, ARCA Services Inc., and Connexx Services Inc., on the other hand, dated February 19, 2021 [filed as 10.1 to the Company’s Form 8-K filed on February 25, 2021 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.1 |
|
Articles of Incorporation of Appliance Recycling Centers of America, Inc. [filed as Exhibit 3.3 to the Company’s Form 8-K filed on March 13, 2018 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.2 |
|
Articles of Conversion [filed as Exhibit 3.1 to the Company’s Form 8-K filed on March 13, 2018 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.3 |
|
Articles of Conversion [filed as Exhibit 3.2 to the Company’s Form 8-K filed on March 13, 2018 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.4 |
|
Certificate of Correction to Articles of Incorporation [filed as Exhibit 3.1 to the Company’s Form 10-Q for the quarterly period ended June 30, 2018 (File No 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.5 |
|
Certificate of Change [filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on April 22, 2019 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.6 |
|
Certificate of Correction to Articles of Incorporation of Appliance Recycling Centers of Amercia, Inc. [filed as Exhibit 3.7 to the Company’s Current Report on Form 8-K filed on June 24, 2019 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.7 |
|
Certificate of Designation of Powers, Preferences, and Rights of Series A-1 Convertible Preferred Stock of Appliance Recycling Centers of America, Inc. [filed as Exhibit 3.8 to the Company’s Current Report on Form 8-K filed on June 24, 2019 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.8 |
|
Amended and Restated Certificate of Designation of the Preferences, Rights, and Limitations of the Series A-1 Convertible Preferred Stock of JanOne Inc., dated October 1, 2020 [filed as Exhibit 3.8(a) to the Company’s Current Report on Form 8-K filed on October 2, 2020 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.9 |
|
Articles of Incorporation of JanOne Inc. (the Name Change Subsidiary), filed with the Secretary of State of the State of Nevada on September 6, 2019 [filed as Exhibit 3.9 to the Company’s Current Report on Form 8-K filed on September 13, 2019 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.10 |
|
Certificate of Amendment to Articles of Incorporation, filed with the Secretary of State for the State of Nevada on November 5, 2020 [filed as 3.9 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 26, 2020 filed on November 10, 2020 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.11 |
|
Articles of Merger for JanOne Inc. into Appliance Recycling Centers of America, Inc., filed with the Secretary of State of the State of Nevada on September 9, 2019, and effective on September 10, 2019 [filed as Exhibit 3.10 to the Company’s Current Report on Form 8-K filed on September 13, 2019 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
3.12 |
|
Bylaws of Appliance Recycling Centers of America, Inc. [filed as Exhibit 3.4 to the Company’s Form 8-K filed on March 13, 2018 (File No. 0-19621) and incorporated herein by reference]. |
77
|
|
|
|
|
3.13 |
|
First Amendment to Bylaws of Appliance Recycling Centers of America, Inc. [filed as Exhibit 3.1 to the Company’s Form 8-K filed on December 31, 2018 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
4.1+ |
|
|
|
|
|
|
|
4.2 |
|
Specimen Stock Certificate [filed as Exhibit 4.2 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 26, 2020 filed on November 10, 2020 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.1X |
|
Patent and Know How License Agreement dated November 19, 2019, by and among JanOne Inc., and UAB Research Foundation, TheraVasc, Inc., and the Board of Supervisors of Louisiana State University and Agricultural and Mechanical College, acting on behalf of LSU Health Sciences Center at Shreveport [filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on November 25, 2019 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.2 X |
|
Master Agreement for Development, Manufacturing and Supply Services dated February 5, 2020 by and between JanOne Inc. and CoreRx Inc. [filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on February 7, 2020 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.3 |
|
Promissory Note between JanOne Inc., as the borrower, and Texas Capital Bank, N.A., as lender [filed as Exhibit 10.1 to the Company’s Current Report on Quarterly Report on Form 10-Q filed on August 10, 2020 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.4 |
|
Amended and Restated Promissory Note, effective April 1, 2018, issued by ApplianceSmart Holdings LLC [filed as Exhibit 10.1 to the Company’s Form 8-K filed on December 31, 2018 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.5 |
|
Security Agreement dated December 26, 2018 by and between ApplianceSmart Holdings LLC and Appliance Recycling Centers of America, Inc. [filed as Exhibit 10.2 to the Company’s Form 8-K filed on December 31, 2018 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.6 |
|
Security Agreement dated December 26, 2018 by and between ApplianceSmart, Inc. and Appliance Recycling Centers of America, Inc. [filed as Exhibit 10.3 to the Company’s Form 8-K filed on December 31, 2018 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.7 |
|
Security Agreement dated December 26, 2018 by and between ApplianceSmart Contracting Inc. and Appliance Recycling Centers of America, Inc. [filed as Exhibit 10.4 to the Company’s Form 8-K filed on December 31, 2018 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.8 |
|
Subordination Agreement, dated March 15, 2019, from Appliance Recycling Centers of America, Inc. to Crossroads Financing, LLC [filed as Exhibit 10.1 to the Company’s Form 8-K filed on March 21, 2019 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.9 |
|
Intercreditor and Subordination Agreement, dated March 18, 2019, by and between Appliance Recycling Centers of America, Inc. and Crossroads Financing, LLC [filed as Exhibit 10.2 to the Company’s Form 8-K filed on March 21, 2019 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.10 |
|
Secured Revolving Line of Credit Promissory Note [filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on August 30, 2019 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.11 |
|
Amendment to Secured Line of Credit Promissory Note dated August 25, 2020 between ARCA Recycling, Inc. and Isaac Capital Group, LLC [filed as Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q filed on November 10, 2020 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.12+ |
|
|
|
|
|
|
|
10.13 |
|
Securities Purchase Agreement dated November 8, 2016, between Energy Efficiency Investments, LLC and the Company [filed as Exhibit 10.1 to the Company’s Form 10-Q for the quarter ended October 1, 2016 (File No. 0-19621) and incorporated herein by reference] |
78
|
|
|
|
|
10.14 |
|
Termination Agreement by and between Energy Efficiency Investments, LLC and JanOne Inc. [filed as 10.18 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 28, 2019 filed on April 6, 2020 (File No. 0-19621) and incorporated herein by reference] |
|
|
|
|
|
10.15 |
|
Form of 3% Original Issue Discount Senior Convertible Promissory Note issuable under Securities Purchase Agreement dated November 8, 2016, between Energy Efficiency Investments, LLC and the Company [filed as Exhibit 10.2 to the Company’s Form 10-Q for the quarterly period ended October 1, 2016 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.16 |
|
Form of Common Stock Purchase Warrant issuable under Securities Purchase Agreement dated November 8, 2016, between Energy Efficiency Investments, LLC and the Company [filed as Exhibit 10.3 to the Company’s Form 10-Q for the quarter ended October 1, 2016 (File No. 0-19621) and incorporated herein by reference]. |
|
|
|
|
|
10.17* |
|
2011 Stock Compensation Plan [filed with the Company’s Schedule 14A on March 31, 2011 and incorporated herein by reference]. |
|
|
|
|
|
10.18* |
|
2016 Equity Incentive Plan [filed as Exhibit 10.3 to the Company’s Form 10-K for the fiscal year ended December 31, 2016 (File No. 0-19621) and incorporated herein by reference] |
|
|
|
|
|
10.19* |
|
First Amendment to the JanOne Inc. 2016 Equity Incentive Plan [filed with the Company’s Schedule 14A on October 2, 2020 and incorporated herein by reference] |
|
|
|
|
|
10.20*× |
|
|
|
|
|
|
|
21.1+ |
|
|
|
|
|
|
|
23.1+ |
|
Consent of WSRP LLP, Independent Registered Public Accounting Firm. |
|
|
|
|
|
31.1+ |
|
Certification by Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
|
31.2+ |
|
Certification by Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
|
32.1† |
|
|
|
|
|
|
|
32.2† |
|
|
|
|
|
|
|
101+ |
|
The following materials from our Annual Report on Form 10-K for the fiscal year ended December 29, 2018, formatted in Extensible Business Reporting Language (XBRL): (i) the Consolidated Balance Sheets, (ii) the Consolidated Statements of Operations and Comprehensive Income, (iii) the Consolidated Statements of Cash Flows, (iv) the Consolidated Statements of Shareholders’ Equity, (v) the Notes to Consolidated Financial Statements, and (vi) document and entity information. |
|
|
|
|
|
* |
|
Items that are management contracts or compensatory plans or arrangements required to be filed as an exhibit pursuant to Item 14(a)3 of this Form 10-K. |
|
|
|
|
|
+ |
|
Filed herewith. |
|
|
|
|
|
† |
|
Furnished herewith. |
|
|
|
|
|
× |
|
Portions of this exhibit have been redacted in compliance with Regulation S-K Item 601(b)(10)(iv) |
79
Pursuant to the requirements of Section 13 or Section 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on our behalf by the undersigned, thereunto duly authorized.
|
March 30, 2021 |
JANONE INC. |
|
|
|
|
|
|
|
By |
/s/ Tony Isaac |
|
|
|
Tony Isaac |
|
|
|
Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
Principal Executive Officer |
|
|
|
|
|
/s/ Tony Isaac |
|
Chief Executive Officer |
|
March 30, 2021 |
|
Tony Isaac |
|
|
|
|
|
|
|
|
|
|
|
Principal Financial and Accounting Officer |
|
|
|
|
|
/s/ Virland A. Johnson |
|
Chief Financial Officer |
|
March 30, 2021 |
|
Virland A. Johnson |
|
|
|
|
|
|
|
|
|
|
|
Directors |
|
|
|
|
|
/s/ Tony Isaac |
|
Director |
|
March 30, 2021 |
|
Tony Isaac |
|
|
|
|
|
|
|
|
|
|
|
/s/ Richard Butler |
|
Director |
|
March 30, 2021 |
|
Richard Butler |
|
|
|
|
|
|
|
|
|
|
|
/s/ John Bitar |
|
Director |
|
March 30, 2021 |
|
John Bitar |
|
|
|
|
|
|
|
|
|
|
|
/s/ Nael Hajjar |
|
Director |
|
March 30, 2021 |
|
Nael Hajjar |
|
|
|
|
80